Deficiencia somatotrófica en niños: características clínicas y bioquímicas según la etiología en 75 casos
H. García1, I. Arteche2, V. Mericq2, E. Codner2, A. Ávila2, C. Henríquez2, M. Eggers2, A. Cortinez2, T. Salazar2, P. Carvallo3, R. Roman2, F. Ugarte4, G. Iñiguez2, F. Beas2, F. Cassorla2.
Somatotrophic deficiency in children. Report of 75 cases
¹ Clínica Santa María. Universidad de los Andes
2 Instituto de Investigaciones Materno Infantil (IDIMI), Hospital San Borja Arriarán.
3 Departamento de Biología Celular y Molecular, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile
4 Hospital Exequiel González Cortés. Universidad de Chile.
Recibido el 2 de octubre, 2007. Aceptado el 14 de noviembre, 2007.
Background: Somatotrophic deficiency (SDMT) can be due to a deficiency of growth hormone releasing hormone (GHRH), growth hormone (GH) or insulin like growth factor I (IGF-I). Although its clinical features have been thoroughly described, the diagnosis is still controversial. Now there is an effective treatment with GH or IGF-I for these patients. Aim: To analyze the main clinical, etiological and laboratory characteristics of 75 SD patients (44 males), aged 9.4 + 4.5 years, with severe growth retardation. The diagnosis was confirmed by the lack of response to two GH stimulation tests (Clonidine, Glugagon or Insulin) and low levels of IGF-I or insulin-like growth factor binding protein- 3 (IGFBP-3). Results: In 34 patients (46 %), the cause of DSMT was considered idiopathic (DSMT-I), in 31 (41 %) there was an organic cause (DSMT-O), most commonly caused by malformations or pituitary tumors and in 10 (13 %), it was genetic (DSMT-G) (three patients with Laron’s Syndrome, five with mutations of GH gene and 2 with probable mutations of Prop-1 and Pit-1 genes). IGF-1 levels, were significantly lower in DSMT-O and DSMT-G than in DSMT-I (21.2± 46.1, 23.4±30.3 ng/mL and 50.2 ± 48.3 ng/mL, respectively). The lowest height score corresponded to DSMT-G, compared to DSMT-O and DSMT (–5.7 ± 0.9, -4.0±1.6 and –4.3±1.2 DS, respectively) Conclusions: The high percentage of organic and genetic etiologies in our patients can be due to the systematic search of these diseases. DSMT-G (Laron, mutations in GH and Pit-1 genes) had the most severe growth retardation.
El retraso marcado de crecimiento constituye un desafío diagnóstico y terapéutico en la endocrinología infantil. Las etiologías son tan variadas que involucran prácticamente a toda la pediatría. Uno de los diagnósticos posibles es la deficiencia somatotrófica (DSMT), caracterizada por retraso severo de la talla, retardo de la velocidad de crecimiento y niveles bajos de GH y/o IGF-1. Este cuadro es poco frecuente y su tratamiento complejo y costoso, por lo que estos casos tienden a concentrarse en centros especializados.
La DSMT puede derivar de alteraciones del eje somatotrófico, desde el hipotálamo hasta los receptores hormonales en los cartílagos de crecimiento. De esta forma, si la falla es hipotalámica no se produce el factor liberador de GH (GHRH); si ella radica en la hipófisis, no se secreta suficiente GH y si está asentada en los tejidos periféricos se manifiesta como insensibilidad de ellos a la GH.
A su vez, todos estos defectos pueden ser de naturaleza genética o adquirida y corresponder a defectos de las propias hormonas o de sus receptores. (Los distintos niveles del eje somatotrófico que pueden ser afectados se muestran en la Figura 1).
La deficiencia de GH puede ser aislada o acompañada de falencia de otras hormonas hipofisiarias, constituyendo un hipopituitarismo de mayor o menor grado. Los casos en los cuales la única hormona afectada es la GH (deficiencia aislada de GH) son en su mayoría idiopáticos. Un 5-20% corresponden a mutaciones del gen de la GH o del receptor de GHRH1,2 las que pueden ser de expresión autosómica dominante, recesiva o ligadas al cromosoma X.
La insensibilidad a GH, aunque puede presentarse en enfermedades graves, desnutrición, daño hepático o renal, en la mayoría de los casos es de origen genético3,5 por mutaciones del receptor de GH o por defectos posteriores al receptor, denominados en su conjunto como Síndrome de Laron. Asimismo, se han descrito mutaciones en el gen de la IGF-1 que afectan su síntesis o liberación6 y también mutaciones en el receptor y en la unidad ácido lábil encargada de su transporte7.
La incidencia de la DSMT ha sido estimada en 1/3500 a 1/10000 personas dependiendo de la población en estudio8,9.
La DSMT no tratada tiene consecuencias graves tanto en la etapa infantil como en la adulta. En los niños determina una talla final muy baja y en los adultos se puede agregar patología cardiovascular por su acción en el metabolismo lipídico y composición corporal10,11.
Este estudio comunica una serie importante de pacientes pediátricos con DSMT, atendidos en forma consecutiva en nuestra Institución (IDIMI).
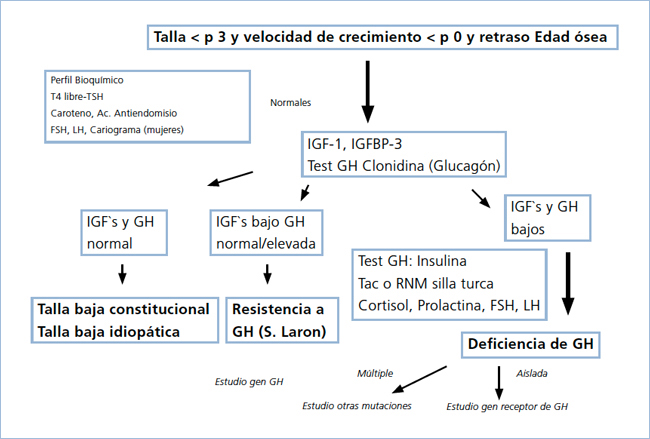
Figura 1. Esquema de las alteraciones en la secreción y acción de GH e IGF.
(Mericq V y Cassorla F 1997. Sistema hormona del crecimiento-efector y su rol en el crecimiento infantil.
Rev. Chil. Pediatr. 68:
27-37.
Reproducido con permiso de los autores y de la Rev.Chil. Pediatr.)
Objetivos
El objetivo del presente estudio fue estudiar las características clínicas y etiológicas de una serie importante de niños chilenos con DSMT, atendidos entre los años 1990 y 2001. También, correlacionar la presentación clínica y la gravedad de las manifestaciones con el tipo etiológico de la DSMT. El diseño corresponde a un estudio de tipo retrospectivo observacional.
Sujetos, Material y Métodos
Unidad de observación: IDIMI, Hospital San Borja Arriarán. Santiago, Chile.
Los 75 pacientes estudiados fueron todos niños derivados desde diferentes regiones de Chile a IDIMI por talla baja grave (<2,5 DS) y deficiente velocidad de crecimiento. Consultaron entre los años 1990 y 2001 y fueron estudiados según un protocolo establecido (Figura 2). En todos ellos se realizó estudio general para descartar otras causas de talla baja, que incluyó examen genético y cariograma en las niñas, perfil bioquímico, evaluación tiroidea y medición de anticuerpos anti-endomisio. Se descartaron los pacientes con Síndrome de Turner o sus variantes.
Los 75 pacientes (44 varones y 31 mujeres) tenían 9,4+4,5 años de edad promedio (0,8 a 21,3 años) y fueron catalogados como portadores de DSMT (Tabla 2). El diagnóstico se sospechó por déficit grave de talla sumado a características clínicas propias de la deficiencia somatotrófica (facies infantil, frente prominente, hipoplasia nasal, voz aguda, microgenitalismo e historia de hipoglicemia) y se confirmó mediante dos pruebas de estímulo de GH, una de ellas con clonidina (en mayores de 3 años) o glucagón (menores de 3 años), y la otra con insulina, en las cuales la respuesta máxima al estímulo fue menor a 7ng/mL, asociado a niveles bajos de IGF-1 y/o IGFBP-3 (Figura 2).
Una vez establecido el diagnóstico de DSMT, se realizó estudio por imágenes de la región hipotálamo-hipofisiaria (TAC o RNM) para descartar etiología orgánica. En los casos con deficiencia de GH aislada, sin anormalidades en el estudio de imágenes selares, se efectuó estudio molecular del gen de GH. Las mutaciones en este gen se identificaron mediante aislamiento del DNA genómico de linfocitos periféricos, usando Southern Blot y amplificación por PCR, digestión enzimática para identificar algunas mutaciones conocidas y secuenciación posterior. La mutación descrita del gen Pit-1 y la del Prop-1 se sospechó en dos casos que presentaban conjuntamente deficiencia de GH, TSH y Prolactina. El análisis molecular de estos genes no ha sido realizado hasta la fecha. En los casos de insensibilidad a GH (Síndrome de Laron) se investigaron posibles mutaciones en el receptor de GH, mediante estudios realizados en el National Institute of Health, en Estados Unidos, y en nuestro centro, según la técnica descrita12.
La DSMT se clasificó como de origen orgánico (DSMTO) si había alteraciones en el estudio radiológico de la región selar. Se consideró de origen genético (DSMT-G) comprobar mutación en el gen de GH o de su receptor, y de origen idiopático (DSMT-I), cuando no estuvieron presentes las razones anteriores.
Determinaciones hormonales
Los niveles de GH fueron determinados por RIA con una sensibilidad de 0,8 ng/mL y coeficientes de variación (CVs) intraensayo e interensayo de 10% y 6,5%, respectivamente. Los reactivos para la medición de GH fueron donados por el National Hormone and Pituitary Program (human GH-I-3, antihuman GH-2 antisera, human GHRP). Los niveles séricos de IGF-I fueron medidos con un RIA desarrollado localmente13, después de extracción con etanol. La sensibilidad de este método es de 5 ng/ mL y los CVs intra e interensayo de 8,6% y 10,2%, respectivamente.
Los niveles de IGFBP-3 fueron determinados por un ensayo immunorradiométrico (IRMA) comercial de Diagnostic System Laboratories (Webster, TX, EE.UU.). La sensibilidad de este ensayo es 0,1 mg/L, con CVs intra e interensayo de 1,1% y 1,8%, respectivamente. Los resultados se presentan como promedios + DS.
Las diferencias entre los grupos se calcularon con el test t de Student.
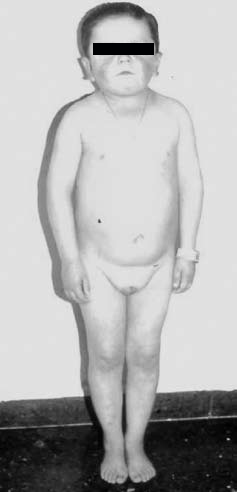
Figura 2. Paciente de 14 años con Síndrome de Laron debido
a mutación del dominio extracelular del receptor de GH.
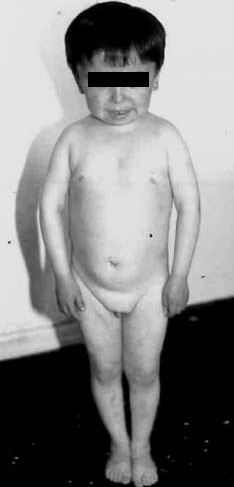
Figura 3. Paciente de 9 años con mutación tipo II a del gen
de GH.
Resultados
En el período de 11 años (1990-2001) en IDIMI se identificaron 75 pacientes con talla baja grave y deficiente velocidad de crecimiento, cuyo estudio permitió catalogarlos como DSMT. La edad cronológica promedio del total fue 9,4+4,5 años y la edad ósea 6,3+3,6 años. El peso corporal y la talla de nacimiento promedios fueron 3144+632 g y 47,8+3,4 cm, respectivamente.
Según el origen de la DSMT se clasificaron como DSMTG, DSMT-O y DSMT-I.
Las características clínicas y antropométricas de los 75 pacientes se muestran en la Tabla 1.
Tabla 1. Características antropométricas de los pacientes con DSMT.Mujeres Hombres Total
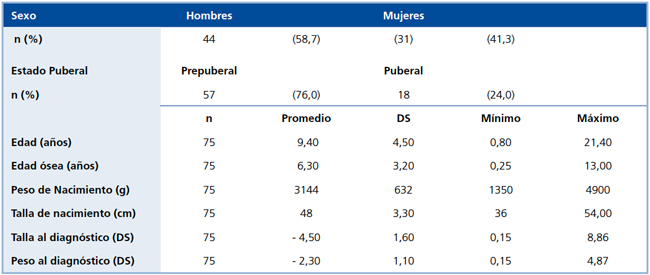
DS: Desviación estándar
Etiología de la deficiencia de GH
De los 75 pacientes estudiados, en 34 (46%) la deficiencia de GH se consideró DSMT-I. En 31 (41%) fue orgánica (DSMT-O) y en 10 (13%) genética (DSMT-G). Al momento del diagnóstico hubo compromiso de otras hormonas hipofisiarias en 6 de 34 con DSMT-I y en 12 de 31 y 2 de 10 de los catalogados como DSMT-O y DSMT-G, respectivamente, siendo la más frecuente el déficit de TSH. Sólo 4 de 31 niños con DSMT-O presentaron diabetes insípida.
Entre las causas orgánicas, las más frecuentes correspondieron a alteraciones del desarrollo de la hipófisis (12 casos, incluyendo 6 pacientes con silla turca vacía y/o aracnoidocele selar), seguidas por 9 casos de tumores del SNC (4 craneofaringeomas, 2 meduloblastomas. y 1 ependimoma y 1 retinoblastoma), 2 secundarios a irradiación del SNC y 2 casos con interrupción del tallo hipofisiario.
En cuanto a las causas genéticas compartieron similar frecuencia el Síndrome de Laron (3 casos) y la mutación del gen de GH (4 casos). La insensibilidad a GH (Síndrome de Laron) incluyó una mutación puntual en el gen del receptor de GH con deleción de 2 bases, 118 y 119, en el exón 4, que corresponde al dominio extracelular del receptor14 (Figura 2) y dos casos de una mutación en el codón 180 del exon 6 del gen del receptor de GH, que introduce un nuevo sitio de splicing15, que corresponde a la misma mutación descrita en los enanos de Loja, Ecuador, la serie más numerosa publicada (56 sujetos afectados)4.
Los niños con mutaciones en el gen de GH corresponden a deficiencia tipo 1a (2 casos) por una mutación de 7 kb y 3 a deficiencia tipo 2 por dos mutaciones diferentes, en la primera y la otra en la quinta base de una zona de splicing del intron 3, resultando ambas en pérdida del exon 3 de la GH16,17 (Figura 3). A pesar de no contar aún con estudio molecular, dos pacientes fueron incluidos como DSMT-G dada la alta sospecha clínica de déficit del Pit-1, al presentar, concomitantemente, disminución de prolactina y tiroxina (Tabla 4).
Al correlacionar las características antropométricas y bioquímicas de los pacientes con deficiencia de GH con la etiología, se observó un mayor compromiso de talla en las DSMT-G (-5,7± 0,9 DS) con respecto a las DSMTO y DSMT-I (-4,0±1,6 DS y -4,3±1,2 DS, respectivamente). Asimismo, las DSMT-G y DSMT-O presentaron valores de IGF-1 de 21,2 ± 46,1ng/mL y 23,4±30,3 ng/ mL, respectivamente, los cuales a pesar de su gran variabilidad, fueron significativamente menores que en los casos idiomáticos, cuyo valor fue 50,2 ± 48,3 ng/mL.
Los niños con probable mutación en el gen de Pit-1 fueron los que tuvieron valores más bajos de GH e IGFBP-3 (0,5+0,5 ng/dL y 0,5+0,1 mg/L, respectivamente), mientras que los menores valores de IGF-1 se observaron en los casos de mutación del gen de GH (Tabla 5).
Los pacientes con insensibilidad a GH eran los de mayor edad al momento del diagnóstico (16,8+4,1 años), mayor compromiso de talla (-7,4+2,0 DS) y de peso (-3,5+1,6 DS), además de presentar niveles de GH muy por sobre los valores normales, lo cual es propio de este diagnóstico. Estos pacientes tenían fenotipo característico con macrocefalia relativa, abombamiento frontal, adiposidad central y escleras oculares azules, elemento clínico descrito en el Síndrome de Laron y en la deficiencia de Prop-1.
Los pacientes con mutación del gen de GH y los con probable mutación del gen Pit-1, presentan un fenotipo muy similar, sin escleras azules, agregándose microgenitalismo. La mayoría de los DSMT-I presentaban únicamente adiposidad central y facies infantil.
Respuesta máxima de GH a las pruebas de estimulación
En todos los pacientes la respuesta a las pruebas de estimulación estuvo muy por debajo de lo considerado normal (>7 ng/dL). En los 57 niños en edad prepuberal el valor promedio de GH después del estímulo fue 1,9+1,5 ng/dL, mientras que en los 18 prepúberes fue 3,5+1,5 ng/dl (Tabla 2). Aunque en los prepuberales no se hizo impregnación con esteroides sexuales, los niveles extremadamente bajos de GH hacen poco probable la presencia de falsos positivos.
Sensibilidad de IGF-1 e IGFPB-3
La correlación entre IGFPB-3 e IGF-1 y las pruebas de estímulo fue alta, de tal manera que la sensibilidad de IGF-1 fue 95,2% ó 86,4% (dependiendo si el punto de corte se aplicaba para los valores de IGF-1, en -1 DS o -2 DS, respectivamente). La sensibilidad de IGFBP-3 fue de 90,5% ó 95,2%, aplicando el mismo criterio. Al considerar ambas en conjunto, la sensibilidad aumentó a 100% (Tabla 3).
Tabla 2. Respuesta máxima de GH en los test de estimulación en pacientes con DSMT.
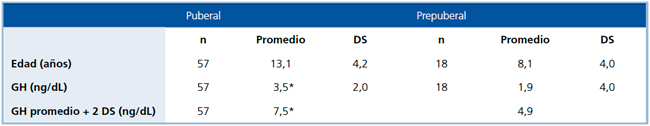
DS: Desviación estándar
*p<0.05
Tabla 3. Sensibilidad de IGF-1 e IGFBP-3.
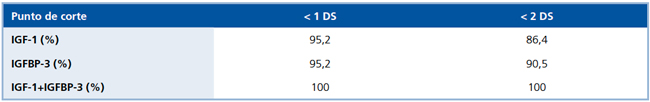
DS: Desviación estándar
Tabla 4. Etiología de la deficiencia somatotrófica (DSMT) en 75 casos (199 0-199 8).
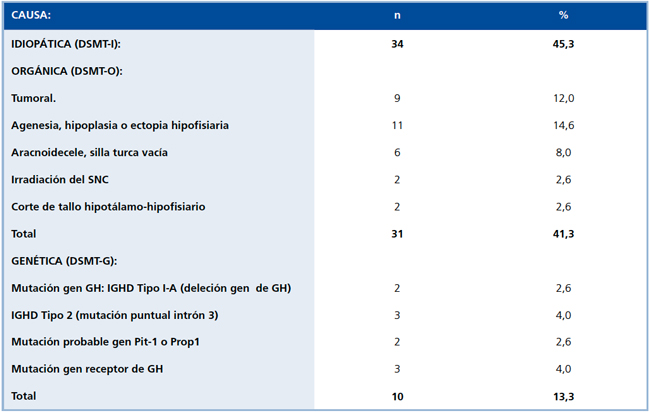
DS: Desviación estándar
Tabla 5. Etiología de la deficiencia somatotrófica (DSMT) en 75 casos (199 0-199 8).
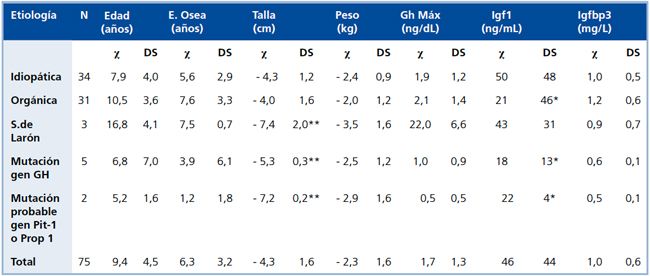
* p<0.05 con respecto a etiología Idiopática
** p<0.05 con respecto a etiología Idiopática y Orgánica
El cálculo excluye pacientes con Síndrome de Larón
Discusión
Este trabajo analiza las características clínicas, bioquímicas y etiológicas de una serie numerosa de pacientes chilenos con DSMT que fueron atendidos en el curso de la última década en IDIMI. Destaca la alta incidencia de causas orgánicas y genéticas en su etiología. Las DSMT-G presentan un fenotipo peculiar con más grave compromiso de talla, abombamiento frontal y microgenitalismo. Asimismo, sus niveles de IGF-1 son los más bajos. La DSMT-O se acompaña más frecuentemente de compromiso de otras hormonas hipofisiarias, mientras que la DSMT-I corresponde mayoritariamente a déficit aislado de GH.
Todos nuestros casos presentaban una clara deficiencia de GH, aun considerando que los métodos para establecer tal déficit son controvertidos18, ya que la estimación de la secreción de GH no es precisa. En este sentido, ninguna prueba de provocación simula satisfactoriamente el patrón normal de secreción de GH19, además que las definiciones de respuestas normales o anormales varían según los diferentes centros de estudio (valor máximo normal de GH entre 5 y 10 ng/dL)20, y dependen de la edad. También hay que considerar que las concentraciones de esteroides sexuales influyen en los niveles de GH, como se demuestra en niños prepuberales que, no respondiendo a las pruebas de provocación, lo hacen normalmente luego de un breve tratamiento con esteroides sexuales21,22. Por último, la reproducibilidad de las pruebas de provocación es pobre23,24.
Así, el diagnóstico de DSMT no puede basarse exclusivamente en las determinaciones de GH y debe considerar el contexto del cuadro clínico del paciente, el registro cuidadoso de la curva de crecimiento, la maduración ósea, los antecedentes familiares, y la determinación de los niveles de IGF-1 e IGFBP-3.
Todos nuestros pacientes presentaban valores de GH muy por debajo de los considerados normales tanto para la etapa prepuberal como para la puberal. Estas concentraciones se correlacionaron con la gravedad del compromiso antropométrico y de IGF-1 e IGFBP-3. Es así que los pacientes con DSMT-G presentaron el déficit de talla más pronunciado, junto a los niveles más bajos de GH, IGF-1 e IGFBP-3, coincidente con lo comunicado en la literatura25 y permitiendo hacer un diagnóstico precoz en la mayoría de los casos.
El hecho de que en nuestros pacientes prepuberales no hiciéramos impregnación con esteroides sexuales antes de las pruebas de estímulo para GH, pudiera hacer plantear la posibilidad de falsos positivos, sin embargo, el cuadro clínico característico y los niveles de GH extremadamente bajos hacen que esto sea poco probable.
Sobre la base de los resultados de las pruebas de estímulos de nuestros pacientes y la alta sensibilidad de IGF-1 e IGFBP-3, hecho demostrado por nuestro grupo26, pudimos establecer un punto de corte para las pruebas de estímulo en el diagnóstico de deficiencia de GH, el que fue 5 ng/dL en pacientes prepuberales (sin impregnación previa) y 7,5 ng/dL en los puberales, valor que se obtiene de sumar 2 DS al promedio de las determinaciones obtenidas.
En cuanto a la sensibilidad de IGF-1 e IGFBP-3 para el diagnóstico de DSMT, ésta fue similar al considerar como punto de corte <1 DS (95%). No obstante, la sensibilidad era mayor para IGFBP-3 si se tomaba como corte <2 DS. Lo anterior se puede explicar porque la IGFBP3 a diferencia de IGF-1, varía menos con la edad, es menos dependiente del estado nutricional y tiene una clara relación con los niveles de GH28,29. Al considerar ambas mediciones en conjunto, la sensibilidad sube a 100%, lo que apoya su determinación en el estudio de pacientes con sospecha de DSMT. La gran sensibilidad encontrada para IGF-1 e IGFBP-3 es mayor que la descrita en otros estudios27-30, lo que puede explicarse por la gravedad de la deficiencia de GH observada en nuestra población.
La mayor parte de los casos de DSMT comunicados en la literatura son de origen idiopático (60-70%), pero cada vez más en este grupo se reconocen mutaciones que afectan distintos locus, como el gen del GHRH y su receptor el gen de GH, o a factores de trascripción (mutaciones en el gen del HESX1, LHX3, Prop-1 y Pit-1)25, 31-35.
Las causas orgánicas que afectan el sistema nervioso central corresponden a 20-25% de los casos. Las más frecuentes son las alteraciones de la anatomía hipofisiaria, la hipoplasia y disrupción del tallo hipotálamo-hipofisiario36 (hoy posibles de diagnosticar con Resonancia Nuclear Magnética), y las tumorales (craneofaringiomas, germinomas, gliomas, neurofibromas)37,38. Además, incluyen algunas malformaciones cerebrales (holoprosencefalia y displasia septo-óptica)39,40, traumatismos (perinatales u otros)36, infecciones (encefalitis viral, bacteriana o fúngica)41; hipofisitis autoinmune42 e irradiación cerebral43, esta última cada vez más frecuente en niños que sobreviven al cáncer.
En la literatura las causas de origen genético fluctúan entre 5% y 10%, aunque con el descubrimiento de nuevas mutaciones aumentará la importancia de este grupo.
Al analizar los porcentajes correspondientes a las diferentes etiologías de DSMT (46% SDMT-I, 41% DSMT-O y 13%DSMT-G) y compararlos con las series presentadas en Norteamérica, Europa y Japón44, en nuestra serie se constata mayor porcentaje de causas orgánicas y genéticas. August y colaboradores publicaron una serie numerosa de pacientes con deficiencia de GH, de los cuales el 72% correspondió a causa idiopática y sólo el 23% a causa orgánica45. Cabe señalar que muchos de los pacientes incluidos en dicho estudio no cumplirían con nuestros criterios de inclusión. La magnitud de la etiología genética publicada varía entre 5% y 10%.
La mayor incidencia de etiología orgánica y genética que describimos puede deberse a la estrictez de los criterios diagnósticos, así como a la búsqueda sistemática de estas patologías. La incidencia de causas orgánicas en nuestra serie puede estar sobredimensionada por la inclusión de casos con aracnoidocele selar y silla turca vacía, los cuales también pueden considerarse como variantes anatómicas. La gran definición de las imágenes de RNM de hipófisis e hipotálamo favorecen una mejor aproximación anatómica a las DSMT, de manera que en muchas insuficiencias de GH consideradas idiopáticas, la RNM puede ahora revelar anomalías en la región selar. Así, Fujisawa46 describió en 1987 los primeros casos de interrupción del tallo pituitario (ITP) en niños con enanismo hipofisario idiopático, manifestado como desaparición del tallo o al menos no visualización de él, (señal no identificada tras el gadolinium), hipófisis anterior hipoplásica y neurohipófisis ausente o ectópica. Posteriormente, numerosos autores realizaron estudios de RNM en niños con deficiencias idiopáticas, encontrando ITP en 20% a 40 % de los casos, frecuentemente asociadas a hipoplasia de la anterohipofisis y a neurohipófisis ectópica47,48. Parece existir una cierta relación entre la forma completa de este síndrome de tallo no visualizado o interrumpido y la forma incompleta constituida por un tallo filiforme, de modo que ésta podría corresponder a una variante atenuada de la primera49.
Estudios recientes sugieren que así como casi todos los casos de tallo “adelgazado” se traducen en deficiencias aisladas de GH, las ITP completas están ligadas a panhipopituitarismo50. La etiopatogenia de este síndrome permanece sin aclarar. La hipótesis traumática, como podría ser el nacimiento por fórceps y la vascular (isquemia), va perdiendo terreno a favor de una hipótesis genética (varios casos en una misma familia, predominio masculino y mayor asociación a malformaciones cerebrales: 12% versus 7% en los con déficit de GH con RNM normal)51. Sólo la RNM practicada sistemáticamente en pacientes con déficit de GH parcial o completo, aislado o múltiple, puede confirmar o rechazar la existencia de esta anomalía. En el caso de ITP, si el déficit de GH es aislado, es necesario el seguimiento de los pacientes, ya que otras deficiencias hipofisarias pueden aparecer en el curso de la observación.
En resumen, una alta incidencia de anormalidades hipofisiarias está presente en el déficit idiopático de GH; los compromisos deficitarios hormonales son más dependientes del tipo de anormalidad hipofisiaria que del tamaño de la hipófisis. La hipoplasia hipofisiaria y la interrupción del tallo, junto a la ectopia de la neurohipófisis deberían ser consideradas como una entidad diferente que refleja un compromiso precoz del desarrollo del hipotálamo e hipófisis.
La mayoría de los casos en nuestra serie se estudiaron con TAC y los menos con RNM. Es probable que si todos hubiesen sido estudiados con RNM de última generación hubiera aumentado aún más la incidencia de trastornos orgánicos hipotálamo-hipofisiarios.
Sobre la base de nuestros resultados, que demuestran un significativo número de casos de origen genético, sugerimos ampliar los estudios moleculares en búsqueda de deleciones o mutaciones del gen de GH en el caso de deficiencia aislada de GH o de Prop-1, Pit-1, HESX o LHX3, o en casos de deficiencias hormonales múltiples, los señalados factores de trascripción. Estos factores se expresan en la hipófisis según un calendario secuencial muy preciso, durante el cual el ectodermo oral, inicialmente invaginado hacia el sistema nervioso central, se diferencia en distintas líneas celulares. El primero en aparecer es el HESX 1 que inicia la diferenciación hipofisiaria y de los nervios ópticos. Su mutación se relaciona con el Síndrome de Displasia Septo Óptica, caracterizado por deficiencias hormonales hipofisiarias múltiples, micro o anoftalmía y ausencia de cuerpo calloso y/o septum pellucidum25. El LHX3 es un factor de trascripción que aparece en forma posterior al HESX 1 y su mutación produce ausencia de GH, TSH, Prolactina y Gonadotropinas, sumados a versión anterior y rotación de los hombros34. Las mutaciones del Prop-1 producen deficiencia hormonal que compromete las mismas líneas hormonales anteriores, sumadas a un fenotipo con escleras azules y disminución del ángulo de flexión de los codos. En las series publicadas de deficiencias hormonales múltiples la mutación de este factor es la más frecuente52. Las mutaciones del Pit-1 generan ausencia de GH, Prolactina y TSH; esta última puede manifestarse en forma más tardía34.
El tratamiento de la DSMT con GH humana (extraída de hipófisis en un comienzo, y posteriormente con GH recombinante) o IGF-1 en los casos de insensibilidad a GH, ha demostrado ser exitoso a través de más de cuatro décadas53. Los pacientes pediátricos a los cuales se ha suministrado GH presentan crecimiento acelerado, pudiendo, en teoría, alcanzar su talla diana adulta si el tratamiento se inicia precozmente. Sin embargo, esta meta no se logra en la mayoría de los casos, implicando que existen aspectos del tratamiento con GH que aún pueden ser optimizados54. En la etapa adulta los pacientes no sustituidos con GH tienen mayor riesgo de enfermedades cardiovasculares por alteraciones en la composición corporal, metabolismo lipídico y glucídico. Además, estos sujetos presentan disminución de la masa muscular y de la mineralización ósea y algunas alteraciones en su estado anímico55,56, todo lo cual se puede revertir en un porcentaje significativo administrando GH57.
Dado el alto costo del tratamiento y el bajo nivel económico de la mayoría de nuestros pacientes, es necesario desarrollar un programa a escala nacional que contemple financiar, en los que cumplan los criterios de deficiencia de GH, su estudio y tratamiento. En ese sentido, nuestro trabajo constituye un llamado de atención hacia ese objetivo.
En resumen, se presenta una serie de 75 casos de deficiencia somatotrófica, la mayor publicada en Chile. En ella se analizan las características clínicas y de laboratorio de la DSMT relacionándola con su etiología. La mayoría correspondió a deficiencia idiopática de GH, aunque la incidencia de causas orgánicas y genéticas es mayor que la publicada en otras series.
Referencias
- Phillips JA, Hjell BL, Seeburg PH, Zachmann M 1981. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci USA 78:6372.
- Phillips JA : Genetic defects in processing growth hormone. In Bercu BB(ed): Basic and clinical aspects of growth hormone. New York,Plenum,1988,pp 57-67.
- Rosenfeld RG , Rosenbloom AL, Guevara-Aguirre J 1994. Growth hormone (GH) insensitivyti due to primary GH receptor deficiency. Endocr Rev 15:369-373.
- Guevara-Aguirre J, Rosenbloom AL, Fielder PJ, Diamond FB Jr, Rosenfeld RG1993. Growth hormone receptor deficiency in Ecuador: clinical and biochemical phenotype in two populations. J Clin Endocrinol Metab. Feb;76(2):417-423.
- Berg MA, Argente J, Chernausek S, Gracia R, Guevara-Aguirre J, Hopp M, Pérez-Jurado L, Rosenbloom A, Toledo SP, Francke U 1993. Diverse growth hormone receptor gene mutations in Laron syndrome. Am J Hum Genet. May;52(5):998-905.
- Camacho-Hübner C, Woods KA, Clark AJ, Savage MO 2002. Insulinlike growth factor (IGF)-I gene deletion. Rev Endocr Metab Disord. Dec;3(4):357-361.
- Domené HM, Bengolea SV, Martínez AS, Ropelato MG, Pennisi P, Scaglia P, et al 2004. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med. 2004 Feb 5;350(6):570-577.
- Lindsay R, Feldkamp M, Harris D, Robertson J, Rallison M 1994. Utah growth study: growth standards and the prevalence of growth hormone deficiency. J Pediatr 125:29-35.
- Stochholm K, Gravholt CH, Laursen T, Jørgensen JO, Laurberg P, Andersen M, et al 2006. Incidence of GH deficiency: a nationwide study. Eur J Endocrinol. Jul;155(1):61-71.
- Rodman D, Kutner MH, Rogers CM, Lubin MF, Fleming GA, Bain RP 1981. Impaired growth hormone secretion in the adult patient: relation to age and adiposity. J Clin invest,67:1361-1369.
- Jorgensen JOL, Pedersen SA, Thuesen l, Jørgensen J, Ingemann- Hansen T, Skakkebaek NE et al. 1989. Beneficial effects of growth hormone treatment in GH-deficient adults. Lancet;1:1221-1225.
- Aguirre E, Benítez R, Youlton R, Carvallo P. 1993. Molecular genetic study of a severe growth hormone deficiency in a Chilean family.Rev Med Chile;121(9):982-986.
- Iñíguez G, Villavicencio A, Gabler F, Palomino A, Vega M 2001. Effect of nitric oxide on the expression of insulin-like growth factors and the insulin-like growth factor binding proteins throughout the lifespan of the human corpus luteum. Reproduction; 122: 865-873.
- Counts DR, Cutler GB Jr. 1995. Growth hormone insensitivity syndrome due to point deletion and frame shift in the growth hormone receptor J Clin Endocrinol Metab. Jun;80(6):1978-1981.
- Espinosa C, Sjoberg M, Salazar T, Rodríguez A, Cassorla F, Mericq V, Carvallo P. 2007. E180 Splice mutation in the growth hormone receptor gene, in a chilean family with growth hormone insensitivity:a probable common mediterranean ancestor ( aceptado para publicación en el JCEPM).
- Molina G, Rodriguez A, Derpich M, Missarelli C, Cassorla F, Mericq V, Carvallo P 2003. Isolated growth hormone deficiency in Chilean patients: clinical and molecular analysis. J Pediatr Endocrinol Metab. Oct-Nov;16(8):1143-1155.
- Missarelli C, Herrera L, Mericq V, Carvallo P 1997. Two different 5’ splice site mutations in the growth hormone gene causing autosomal dominant growth hormone deficiency. Hum Genet. Nov;101(1):113- 117.
- Rosenfeld RG, Albertsson-Wikland K, Cassorla F, Frasier SD, Hasegawa Y, Hintz RL et al 1995. Diagnostic controversy: The diagnosis of childhood growth hormone deficiency revisited. J Clin Endocrinol Metab 80:1532-1540.
- Raiti S, Davis WT, Blizzard RM 1967. A comparison of the effects of insulin hypoglycemia and arginine infusion on release of human growth hormone. Lancet 2:1182-1187.
- Physicians’ Desk Reference,48th ed.Montvale, NJ , Medical Economics Data Production Company,1994, p1004.
- Lippe B, Wong S-LR, Kaplan SA 1971, Simultaneous assessment of growth hormone and ACTH reserve in children pretreated with diethylstibesterol. J Clin Endocr 33:949-956.
- Marin G, Domené HM, Barnes KM, Blackwell BJ, Cassorla FG, Cutler GB Jr 1994. The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys. J Clin Endocrinol Metab. Aug;79(2):537- 541.
- Cacciari E, Tassoni P, Parisi G, Pirazzoli P, Zucchini S, Mandini M et al 1992. Tassoni P, Parisi G, et al. Pitfalls in diagnosing impaired growth hormone (GH) secretion: retesting after replacement therapy of 63 children defined as GH deficient. J Clin Endocrinol Metab 74:1284-1289.
- Rakover Y, Lavi I, Masalah R, Issam T, Weiner E, Ben-Shlomo I 2000. Comparison between four immunoassays for growth hormone (GH) measurement as guides to clinical decisions following GH provocative tests. J Pediatr Endocrinol Metab. Jun;13(6):637-643.
- Rimoin DL., Genetic disorders of the pituitary gland. In : Rimoin DL, Connor JM, Pyeritz RE eds. Principle and practice of medical genetics, 3rd ed. 1997 New York. Churchill Livingstone,1331-1364.
- Codner E, Mericq V, Ugarte F, Iniguez G, Espinoza M, Avila A, et al 1999. Usefulness of the measurement of insulin-like growth factor (IGF-I) and IGF-1 binding protein-3 (IGFBP-3) for the diagnosis of growth hormone (GH) deficiency in children]. Rev Med Chile Jul;127(7):807-813.
- Gargosky SE, Pham HM, Wilson KF, Liu F, Giudice LC, Rosenfeld RG 1992. Measurement and characterization of binding protein for insuline growth factor binding protein-3 in human biological fluids: discrepancies between radioimmuno oassay and ligand blotting. Endocrinology 131:3051-3060.
- Baxter RC, Martin JL 1986. Radioimmunoassay of growth hormone dependent insulin-like growth factor binding protein in human plasma. J Clin Invest 78:1504-1512.
- Hasegawa Y, Hasegawa T, Aso T, Kotoh S, Tsuchiya Y, Nose O et al 1992. Usefulness and limitation of mesearement of insuline-like growth binding protein-3(IGFBP-3) for diagnosis of growth hormone deficiency. Endocrinol jpn 39:585-389.
- Smith WJ, Nam TJ, Underwood LE, Busby WH, Celnicker A, Clemmons DR 1993. Use of insulina-like growth factor binding protein- 2(IGFBP-2), IGFBP-3, and IGF-1 for assessing growth hormone secretion. J Clin Endocrinol Metab 77:1294-1299.
- Barinaga M, Yamonoto G, Rivier C, Vale W, Evans R, Rosenfeld MG 1983. Transcriptional regulation of growth hormone gene espression by growth hormone-releasing factor. Nature 306:84-85.
- Esch FS, Böhlen P, Ling NC, Brazeau PE, Wehrenberg WB, Guillemin R 1983. Primary structure of three human pancreas peptides with growth hormone releasing activity. J Biol Chem 258:1806-1812.
- Nelson C, Albert VR, Elsholtz HP, Lu LI, Rosenfeld MG 1988. Activation of cell-specific expression of rat growth hormone and prolactin genes by a common transcription factor. Science 239:1400-1405.
- Li S, Crenshaw EB 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG 1990. : Dwarf locus mutants licking three pituitary cell types result from mutation in the POU domain gene PIT-1. Nature 347:528-533.
- Woodside KJ, Shen H, Muntzel C, Daller JA, Sommers CL, Love PE 2004. Expression of Dlx and Lhx family homeobox genes in fetal thymus and thymocytes. Gene Expr Patterns. May;4(3):315-320.
- Arifa N., Léger J., Garel C., Czernichow P., Maghnie M, Larizza D et al 1991. Hypopituitarism and stalk agenesis: a congenital syndrome worsened by breech delivery? Horm Res.;35(3-4):104-108.
- Costin G 1979. Endocrine disorders associate with tumor of the pituitary and hypothalamus. Pediatr Clin North Am 26:15-31.
- Jenkins JS, Gilbert CJ, Ang V 1976. Hypothalamic-pituitary function in patiens with craniopharyngiomas . J Clin Endocrinol Metab 43: 394- 399.
- Hintz RL, Menking M, Sotos JF 1968. Familial holoprosencephaly with endocrine disgenesys. J Pediatr 72:81-87.
- Hoyt WF, Kaplan SL, Grumbach MM 1970. Septo-optic displasia and pituitary dwarfism. Lancet 1:893-894.
- Bartsocas CS, Pantelakis SN 1973. Human growth hormone therapy in hypopituitarism due to tuberculous meningitis. Acta Paediatr Scand 62:304-306.
- Mayfield RK, Levine JH, Gordon L 1980. Lynphadenoid hypophysitis presenting as a pituitary. Am J Med 69:619-623.
- Brauner R, Rappaport R, Prevot C, Czernichow P, Zucker JM, Bataini P et al 1989. A prospective study of the development of growth hormone deficiency in children given cranial irradiation, and its relation to statural growth. J Clin Endocrinol Metab 68:346-351.
- Wilton P, Wallstrom A 1991. An overview of the diagnoses in the Kabi Pharmacie International Growth Study. Acta Paediatr Scand; Suppl.379:93-98.
- August GP, Lippe BM, Blethen SL, Rosenfeld RG, Seelig SA, Johanson AJ et al 1990. Growth hormone treatment in the United States: demographic and diagnostic features of 2331 children. J Pediatr; 116:899-903.
- Fujisawa I, Kikuchi K, Nishimura K, Togashi K, Itoh K, Noma S et al 1987. Transection of the pituitary stalk: development of an ectopic posterior lobe assessed with MR imaging. Radiology 165: 487-489.
- Ultmann M,C Siegel SF, Hirsch WL, Finegold DN, Foley TP1993. Pituitary stalk and ectopic hyperintense Tl signal on magnetic resonance imaging. Am J Dis Child; 147:647-652.
- Arifa N, Léger J, Garel C, Czernichow P Hassan M 1999. Anomalies malformatives cérébrales associées á l’insuffisance somatotrope chez llenfant : marqueurs majeurs pour le diagnostic , Arch Pedatr;6:14-21.
- Zimmermann A, Schenk JP, Grigorescu Sido P, Pfaffle R, Lazea C, Zimmermann T et al.2007 MRI findings and genotype analysis in patients with childhood onset growth hormone deficiency--correlation with severity of hypopituitarism. J Pediatr Endocrinol Metab. May;20(5):587- 596.
- Pinto G, Netchine I, Sobrier M.L, Brunelle F, Souberbielle J, Brauner R 1997. Pituitary stalk interruption syndrome: a clinical-bíologícal-genetic assessment of its pathogenesis. J Clin Endocrinol Metab; 82:3450- 3454.
- Maghnie M, Triulzi F, Larizza D, Preti P, Priora C, Scotti G et al 1991. Hypothalamic-pituitary dysfunction in growth hormone-deficient patients with pituitary abnormalities. J Clin Endocrinol Metab. Jul; 73(1):79-83.
- Crone J, Pfaffle R, Stobbe H, Prayer D, Gomez I, Frisch H 2002. Familial combined pituitary hormone deficiency caused by PROP-1 gene mutation. Growth patterns and MRI studies in untreated subjects. Horm Res.;57(3-4):120-126.
- Guyda HJ 1999. Four decades of growth hormone therapy for short children: what have we achieved? Review. J Clin Endocrinol Metab. Dec;84(12):4307-4316.
- Savage MO, Cassorla FG, Gluckman PD, Grueters-Kieslich A, Raghupathy P, Silink M et al 2006. Global inequalities in paediatric endocrine practice: statement of minimal acceptable care. Statement from the international societies for paediatric endocrinology. Horm Res.; 65(3):111-113.
- Dean HJ, McTaggart TL, Fish DG, Friesen HG 1985. The educational, vocational, and marital status of growth hormone-deficient adults treated with growth hormone during childhood. Am J Dis Child 139:1105- 1110.
- Rosen T, Bengtsson BA 1990: Premature mortality due to cardiovascular disease in hypopituitarism. Lancet 336:285-288.
- Clayton P, Gleeson H, Monson J, Popovic V, Shalet SM, Christiansen JS 2007. Growth hormone replacement throughout life: Insights into age-related responses to treatment. Growth Horm IGF Res. 17(5):369- 382.