Panhipopituitarismo y diabetes insípida secundarios a hipofisitis linfocitaria en una paciente con trisomia 12p
Andrea Sepúlveda N.¹, Francisca Eyzaguirre C.², Fanny Cortés M.³, Hernán García B.¹
Panhypopituitarism and diabetes insipidus secondary to a lymphocytic hypophysitis in one patient with trisomy 12p
1Centro Endocrinológico Clínica Santa María.
²Becada Endocrinología Infantil IDIMI, Universidad de Chile. Becada SOCHED.
³Laboratorio de Genética y Enfermedades Metabólicas, INTA, Universidad de Chile.
Correspondencia a: Dra. Andrea Sepúlveda N. easn5@vtr.net Fax: 4612153
Recibido el 21 de septiembre, 2007. Aceptado el 9 de noviembre, 2007.
Lymphocytic hypophysitis (LH) is an uncommon inflammatory disease of the hypophysis. It’s female to male
ratio of appearance is 9:1. Pregnant women are more affected during the third trimester of pregnancy or
postpartum. Clinical and radiological presentation can simulate a hypophyseal adenoma. We report a non
pregnant 13 years old adolescent, with a trisomy 12p, with panhypopituitarism, diabetes insipidus and a
selar tumor. It was necessary to differentiate between a germinoma and a LH. The latter was confirmed
with the hypophyseal biopsy.
(Key words: trisomy 12p; lymphocytic hypophysitis; hypopituitarism)
Introducción
La hipofisitis linfocitaria (HL) es una enfermedad inflamatoria poco frecuente, habiéndose descrito alrededor de 160 casos desde la comunicación original en 19621.
Esta condición es más común en mujeres (9:1) y se presenta con mayor frecuencia durante el tercer trimestre del embarazo o el postparto. La edad promedio de presentación es de 34 años en mujeres y 45 años en hombres2,3. Tiene una fuerte asociación con otras enfermedades autoinmunes (25%), especialmente tiroiditis autoinmune. Dada la falta de especificidad de los marcadores6,7, el diagnóstico de certeza se establece sólo con estudio histológico.
La presentación clínica resulta de los efectos de la masa selar, con o sin síndrome tumoral (56%). Cerca del 40% de los casos se presenta con hiperprolactinemia y aproximadamente un 65% tiene insuficiencia adenohipofisiaria. La diabetes insípida (DI) es mucho menos frecuente, alcanzando a un 19%3,5.
El diagnóstico diferencial de una masa selar con hipopituitarismo se hace entre tumores hipofisiarios, que corresponden a más del 80%, y lesiones inflamatorias no neoplásicas que representan un 8 %. En el caso de niños y adolescentes, los tumores funcionantes son muy infrecuentes9.
Comunicamos el caso de una adolescente de 17 años, con trisomía 12p, que se presentó clínicamente con DI, panhipopituitarismo y masa selar, lo que hizo plantear una linfocitis hipofisiaria y requiriendo descartar un germinoma10.
Caso Clínico
Paciente de 17 años de edad producto del primer embarazo de padres jóvenes, sanos, no consanguíneos. Tiene dos hermanos sanos, con estudio cromosómico normal. Su embarazo evolucionó con síntomas de parto prematuro que requirieron tocolíticos y reposo.
El parto fue de término por cesárea por sufrimiento fetal. El peso al nacer fue 4.800 g (GEG) con talla de 51 cm y CC 37 cm. Apgar: 9-9. Evaluada por genetista en período neonatal fue considerada normal, salvo su macrosomía. Reevaluada por genetista a los 10 meses de edad, destacó hipotonía, retraso del desarrollo psicomotor (RDSM) y una conformación craneofacial especial: pelo y cejas ralas, frente amplia, puente nasal plano, micro fosetas en pabellón hélix auricular derecho y en forma de surco en lóbulo auricular izquierdo, fosas nasales anchas y en anteversión, paladar alto con tendencia a úvula bífida. Presentaba también desplazamiento anterior del ano, alteración de surcos palmares de flexión, aumento de círculos en los dedos y asimetría de extremidades inferiores, por lo que se planteó el diagnóstico de Síndrome de Beckwith-Wiedemann, asociado a alguna alteración cromosómica.
El cariograma mostró cariotipo 46, XX, 22q+ (se informó que el segmento extra podría corresponder a 12p) y se sugirió análisis cromosómico de ambos padres. El cariograma del padre mostró cariotipo 46, XY, 9qh+ (variante normal) y el de la madre 46, XX, t (12; 22). Con el estudio anterior se certificó el diagnóstico de trisomía 12p.
La paciente evolucionó con importante RDSM, logrando marcha independiente a los 2 años. A lo largo del tiempo se ha mantenido en programas de educación especial, sin lograr aún leer o escribir.
Presentó su menarquia a los 14 años y por presencia de sangrados menstruales abundantes, sumado a los problemas inherentes al retraso mental, ginecóloga indicó tratamiento desde los 16 años con anticonceptivo oral (levonorgestrel 0,15 + etinilestradiol 0,03mg).
A los 17 años de edad la madre consulta por notar compromiso del estado general, decaimiento, cefalea y polidipsia importante (>4 L/d). Dentro del estudio la RNM cerebral mostró un proceso expansivo mayor a 2 cm, que comprimía el quiasma óptico, sin invasión de los senos cavernosos. Esta imagen se catalogó como macroadenoma hipofisiario intra y supraselar (Figura 1 y Figura 2). En estudio, la imagen T1 sagital sin gadolinio, mostró una silla turca de tamaño normal con un significativo aumento de volumen de la hipófisis, la cual mostró intensidad de señal normal y homogénea. El tallo hipofisiario estaba ascendido y no se reconocía claramente la señal espontánea hiperintensa de la neurohipófisis. En el corte coronal con gadolinio se evidenció claramente la glándula aumentada de tamaño, protruyendo hacia cefálico, con ocupación de la cisterna supraselar y rechazando el quiasma óptico. La impregnación del contraste es difusa y homogénea, sin defectos de captación glandulares que sugieran lesiones focales de tipo adenomatoso o de degeneración quística.
En el estudio endocrinológico destacó: IGF-I:67 ng/mL; Cortisol <1,0 ug/dL; GH: 3,0 ng/mL; Prolactina <2,0 ng/mL; T4 L: 0,5 ug/dL; TSH: 0,2 uUI/mL; FSH: 0,6 mUI/mL; LH <0,8 mUI/mL; E2 <20 pg/mL. Luego de 8 horas de restricción hídrica, la osmolaridad urinaria fue 200 mosmol/L y la plasmática 305 mosmol/L. Estos exámenes resultaron compatibles con panhipopituitarismo con DI.
Previo a la cirugía se agregó al tratamiento con ACO, desmopresina oral, 0,2 mg/en la noche, levotiroxina 100 ug/d y cortisol 25 mg/d, mejorando ostensiblemente su estado de ánimo y desapareciendo la cefalea.
El neurocirujano intentó efectuar una resección transesfenoidal, la que no fue posible por encontrar una masa de consistencia pétrea no resecable. Sólo se logró tomar muestras para estudio histológico e inmunohistoquímico; las tinciones de anticuerpos Pan-T y Pan-B fueron positivas, concluyéndose el diagnóstico de hipofisitis linfocitaria de tipo autoinmune.
Posterior a la cirugía, se mantiene con tratamiento hormonal de reemplazo con normalización de los niveles de T4 (7,3 ug/dL) y cortisol (12,7 ug/dL). La medición de anticuerpos antihipofisiarios, antitiroideos y antiadrenales resultó negativa. Una nueva RNM al séptimo mes de la cirugía, mostró desaparición completa de la masa descrita. (Figura 3 y Figura 4). En estas imágenes se observa una glándula hipófisis de tamaño y morfología normales, con intensidad de señal homogénea. En los cortes sagitales se aprecia tenuemente la hiperintensidad de la señal neurohipofisiaria y no se reconocen lesiones satélites.
Actualmente la paciente tiene 21 años, se encuentra institucionalizada, con un retraso mental severo, casi completamente dependiente en las actividades de la vida diaria y no ha vuelto a presentar sintomatología que sugiera recidiva de la patología.
Discusión
El diagnóstico de HL es complejo en una adolescente y más aún ante la presencia de una masa selar y de una cromosomopatía. La trisomía 12p es una alteración cromosómica de tipo estructural, caracterizada por presentar 3 copias del brazo corto del cromosoma 12, cuyo origen en general es heredado de una translocación balanceada materna o paterna. Generalmente se manifiesta con peso de nacimiento aumentado, macrocefalia, facies plana con pliegues epicánticos, mejillas prominentes, puente nasal ancho, nariz corta, filtrum largo, labio bucal inferior evertido y retraso mental grave, características todas presentes en nuestra paciente. La literatura también describe mayor frecuencia de tumores viscerales sólidos8, pero no está descrita la asociación con la HL.
Ciertos elementos del cuadro clínico de HL orientan al correcto diagnóstico: insuficiencia adenohipofisiaria que aparece durante o luego de un embarazo, déficit aislado de producción hormonal hipofisiaria (ACTH y/o TSH con secreción normal de GH/ Gonadotrofinas) que no sigue el patrón habitual de pérdida de función hipofisiaria y DI acompañando la insuficiencia de la adenohipófisis11. Rara vez se presenta sólo con DI, con compromiso y engrosamiento reducido al tallo hipofisiario. Esta forma de presentación se llama neurohipofisitis infundibular12.
Los exámenes de laboratorio son muy inespecíficos, pudiendo encontrarse anticuerpos antinucleares y factor reumatoideo positivo o VHS ligeramente elevada. Respecto a los anticuerpos antihipófisis, la experiencia actual señala una baja positividad (<30%), dada la escasa sensibilidad de las técnicas de inmunofluorescencia13.
El autoanticuerpo detectado, correspondería a una proteína de 49-KDa:(alpha)-enolasa, de ubicación citosólica y distribución ubicua (a diferencia de la gamma-enolasa que sería la forma neuronal específica). Sólo la presencia de altos títulos se acompañaría de déficit hormonal. No es específico de esta enfermedad y puede estar presente en otras enfermedades autoinmunes14. Incluso existen casos descritos de pacientes con Síndrome de Sheehan que presentan estos anticuerpos15.
La radiología juega un rol insustituible en el diagnóstico, siendo la RNM el examen de elección. Los elementos que se describen con mayor frecuencia son: masa selar con extensión supraselar; aumento homogéneo de intensidad de toda la adenohipófisis con gadolinio; engrosamiento del tallo hipofisiario; pérdida de la señal hiperintensa de la neurohipófisis y engrosamiento de ésta. La silla turca vacía es una manifestación muy poco frecuente y a la fecha sólo existen comunicaciones de casos aislados16.
El diagnóstico de certeza se hace mediante el estudio histológico de la lesión. La histopatología muestra infiltración difusa de células inflamatorias (linfocitos forman folículos linfoides), fibrosis reactiva de grado variable y compromiso principalmente del lóbulo anterior, que puede ser de tipo linfocitario, granulomatoso o xantomatoso17. Asimismo, el estudio histológico permite descartar con mayor certeza el diagnóstico de germinoma. Si bien este tumor no es frecuente en pacientes con trisomía 12p, éste sí puede presentarse con DI y panhipopituitarismo, cuyo tratamiento puede llevar a retrasar el diagnóstico y empeorar el pronóstico del paciente10,18. En este caso, la intervención quirúrgica se planteó como primera opción terapéutica como una forma de aclarar la etiología del panhipopituitarismo y la DI en una paciente con un cuadro clínico muy poco frecuente, asumiendo que era un procedimiento riesgoso y que a largo plazo podría determinar una insuficiencia hipofisiaria permanente.
Esta patología puede evolucionar hacia la recuperación espontánea de la función hipofisiaria, hacia el hipopituitarismo progresivo y permanente o episodios de recurrencia19. De una revisión de 128 casos reunidos en la Clínica Mayo, en 6 de ellos hubo regresión espontánea de la masa. Una de estas mujeres logró un embarazo posterior, sin recurrencia de la lesión en el postparto19. Algunos casos de síndrome de silla turca vacía podrían corresponder a casos de HL que han tenido resolución espontánea.
El tratamiento de esta patología es quirúrgico, ya que la cirugía transesfenoidal es tanto diagnóstica como terapéutica cuando hay extensión supraselar. Sin embargo, dada la evolución variable y considerando que se trata de una enfermedad benigna, ésta debe ser mínimamente invasiva y, por lo tanto, obliga a una fuerte presunción diagnóstica previa4,5.
De lo anterior se puede concluir que todo paciente con HL debe tener un seguimiento endocrinológico y de imágenes a largo plazo, ya que las recurrencias pueden ser tratadas con altas dosis de corticoides antes de plantear una reintervención quirúrgica.
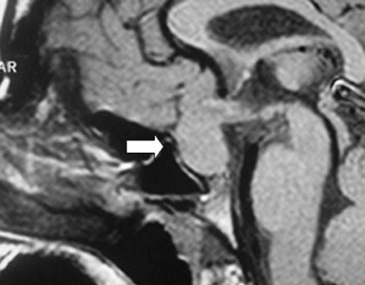
Figura 1. RNM inicial (corte sagital) donde se evidencia la masa selar.
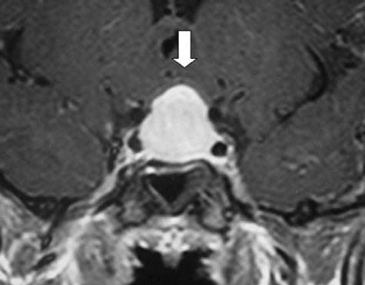
Figura 2. RNM inicial (corte coronal) donde se evidencia la masa selar.
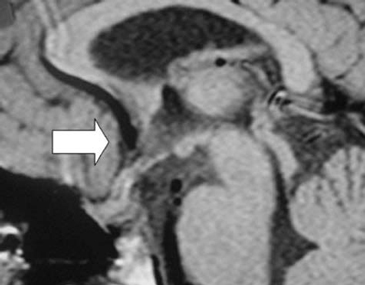
Figura 3. RNM 7 meses después de operada (corte sagital) sin evidencia de la masa selar.
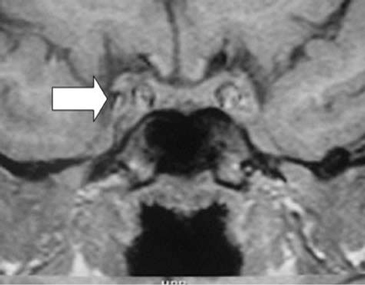
Figura 4. RNM 7 meses después de operada (corte coronal) sin evidencia de la masa selar.
Referencias
- Goudie RB, Pinkerton PH. 1962. Anterior hypophysitis and Hashimoto’s disease in a young woman. J Pathol Bacteriol 83: 584-585.
- Pestell RG, Best JD, Alford FP. 1990. Lymphocytic hypophysitis: the clinical spectrum of the disorder and evidence for an autoimmune pathogenesis. Clin Endocrinol (Oxf) 33(4): 457-466.
- Thodou E, Asa SL, Kontogeorgos G, Kovacs K, Horvath E, Ezzat S. 1995 Clinical case seminar: Lymphocytic hypophysitis: Clinicopathological findings. J Clin Endocrinol Metab; 80(8): 2302-2311.
- Kristof RA, Van Roost D, Klingmuller D, Springer W, Schramm J. 1999. Lymphocytic hypophysitis: non invasive diagnosis and treatment by high dose methylprednisolone pulse therapy? J Neurol Neurosurg Psychiatry 67(3): 398-402.
- Chico A, Puig-Domingo M, Martul P, De Juan M, Prats JM, Mauricio D et al.1998. Reversible endocrine dysfunction and stalk enlargement. J Clin Endocrinol Invest. 21(2): 122-127.
- Ozawa Y, Shishiba Y. 1993. Recovery from lymphocytic hypophysitis associated with painless thyroiditis: clinical implications of circulating antipituitary antibodies. Acta Endocrinol. (Copenh) 128(6):493-498.
- Rivera J A. 2006. Lymphocytic hypophysitis: Disease spectrum and approach to diagnosis and therapy. Pituitary 9(1): 35-45.
- Segel R, Peter I, Demmer LA, Cowan JM, Hoffman JD, Bianchi DW. 2006. The natural history of trisomy 12p. Am J Med Genet A. 140(7): 695-703.
- Scott R, Hedley-Whyte E, Tessa. 2002. Case record of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 35-2002. A nine-year-old girl with cold intolerance, visual-field defects, and a suprasellar tumor. N Engl J Med 347(20): 1604-1611.
- Mikami-T Y, Akiyama M, Yanagisawa T, Takahashi-F. J, Yokoi K, Fukuoka K et al. 2006. Lymphocytic hypophysitis with central diabetes insipidus and subsequent hypopituitarism masking a suprasellar germinoma in a 13-year-old girl. Childs Nerv Syst. 22(10): 1338-1343.
- Cheung CC, Ezzat S, Smyth H, Asa SL. 2001. The spectrum and significance of primary hypophysitis .J Clin Endocrinol Metab 86(3):1048- 1053.
- Maghnie M, Cosi G, Genovese E, Manca-Bitti ML, Cohen A, Zecca S et al. 2000. Central diabetes insipidus in children and young adults.N Engl J Med 343(14):998-1007.
- De Bellis A, Bizzarro A, Conte M, Perrino S, Coronella C, Solimeno S et al.2003. Antipituitary Antibodies in Adults with Apparently Idiopathic Growth Hormone Deficiency in Adults with Autoimmmune Endocrine Diseases. J Clin Endocrinol Metab 88(2): 650-654.
- O’Dwyer DT, Smith AI, Matthew ML, Andronicos NM, Ranson M, Robinson PJ et al. 2002. Identification of the 49 KD-a autoantigen associated with Lymphocytic Hypophysitis an (alpha)-enolase. J Clin Endocrinol Metab 87(2): 752-757.
- . Goswami R, Kochupillai N, Crock PA, Jallel A, Gupta N .2002. Pituitary Autoimmunity in patients with Sheehan’s Syndrome . J Clin Endocrinol Metab 87(9): 4137-4141.
- Unluhizarci K, Bayram F, Colak R, Ozturk F, Selcuklu A, Durak AC et al. 2001. Distinct Radiological and Clinical Appearance of Lymphocytic Hypophysitis. J Clin Endocrinol Metab 86(5): 1861-1864.
- Gutenberg A, Buslei R, Fahlbusch R, Buchfelder M, Brück W. 2005. Immunopathology of primary hypophysitis: implications for patogenesis. Am J Surg Pathol. 29(3):329-38.
- Mootha SL, Barkovich AJ, Grumbach MM, Edwards MS, Gitelman SE, Kaplan SL et al. 1997. Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab. May;82(5):1362-7.
- Gagneja H, Arafah B, Taylor HC. 1999. Histologically proven lymphocytic hypophysitis: spontaneous resolution and subsequent pregnancy. Mayo Clin Proc 74 (2): 150-154.