Enfermedad de Addison en un niño con adrenoleucodistrofia ligada al cromosoma X
Carmen Bustamante E.a, Gloria Durán S.1, Alejandro Martínez A.1, Raúl Escobar H.1, Olga Acuña1 y Hernán García B.1
Primary adrenal failure associated to X- linked Adrenoleukodystrophy. Report of one case
aEstudiante 7º año de Medicina, Escuela de Medicina, Universidad de Antioquia, Colombia.
1División de Pediatría, Escuela de Medicina, Pontificia Universidad Católica de Chile.
Correspondencia:
Dr. Hernán García Bruce
Lira 85. 5º piso, División de Pediatría. Pontificia Universidad Católica de Chile,
Santiago de Chile.
Código postal 8330074
Teléfono 562-3543887
Fax: 562-6384307
E-mail: hgarciab@gmail.com
Recibido: 25 de Mayo de 2010
Aceptado: 14 de Junio de 2010
Primary adrenal failure (PAF) can be congenital or acquired. X- linked Adrenoleukodystrophy (ALD-X), produced by the mutation of the ABDC1 gene (Xq28), that leads to the plasma accumulation of very long chain fatty acids, is one of the congenital diseases associated to adrenal destruction. We report a 7 years old boy with fast progression of right strabismus and general symptoms as weariness, weakness and mucosal and skin pigmentation. A brain magnetic resonance image showed a leukoencephalopathy, characteristic of ALD-X. Low plasma cortisol, high ACTH levels and lack of response to ACTH test, confirmed the diagnosis of primary adrenal insufficiency. High plasma levels of C26:0 fatty acids, and C24/22, C26/22 ratios confirmed ALD-X.
Key words: Adrenal insufficiency, adrenoleukodystrophy, Addison's disease.
La Insuficiencia suprarrenal primaria (ISRP) es una patología infrecuente, con una prevalencia que varía desde 0,45 a 11 casos por 100.000 habitantes. Se presenta con manifestaciones inespecíficas, que exigen del clínico un alto índice de sospecha y explican la tardanza en reconocerla. Según la localización del defecto, la ISR se clasifica en: Primaria (ISRP), cuando se origina en la glándulas suprarrenales, Secundaria (ISRS) cuando es a nivel hipofisiario y Terciaria (ISRT) cuando es hipotalámica1.
La ISRP, también conocida como Enfermedad de Addison, puede tener tanto un origen congénito como adquirido. Clínicamente se manifiesta con astenia y adinamia y molestias gastrointestinales como vómitos y anorexia. También puede presentarse con deshidratación intensa grave que incluso puede producir la muerte si el diagnóstico no es precoz2.
El origen de la Enfermedad de Addison responde a 3 categorías: 1) Disgenesia suprarrenal; 2) Destrucción suprarrenal y 3) Alteración enzimática de la esteroidogénesis suprarrenal3. En la edad pediátrica, la causa más frecuente de ISRP es la Hiperplasia Suprarrenal Congénita (incidencia 1:15.000), mayoritariamente causada por deficiencia de la enzima 21 Hidroxilasa4. Le sigue en frecuencia el origen autoinmune, dentro del contexto del síndrome poliglandular autoinmune (SPG) que ha desplazado a la tuberculosis como principal causa de destrucción de las glándulas suprarrenales. El SPG se asocia a otros daños autoinmunes en más del 45% de los casos, siendo el tiroides el más frecuentemente afectado (> 25%). El SPG tipo 1 comienza generalmente en la primera década de la vida. Para establecer el diagnóstico es suficiente reconocer dos de los tres componentes de la tríada constituida por candidiasis muco cutánea, hipoparatiroidismo e insuficiencia suprarrenal. Pueden también asociarse otros compromisos como hipogonadismo o hipotiroidismo; menos frecuente es la aparición de diabetes. Manifestaciones adicionales como vitiligo, distrofia ungueal, queratopatía, anemia perniciosa o hipoplasia del esmalte dental pueden ocasionalmente estar presentes. La herencia del SPG es de tipo autonómico recesivo ligada a una mutación de un único gen situado en el cromosoma 21q22.3 denominado gen AIRE2-4 (Autoimmune Regulator), mutación de la cual se han descrito más de 40 variedades distintas. Presenta una penetrancia del 100%, sin haberse descrito asociación con HLA respecto a herencia.
Es característica la gran variabilidad en el número y asociación de los componentes del SPG, lo que sugiere factores ambientales que modulan la aparición de la enfermedad. El SPG predomina en el sexo femenino, al igual que la mayor parte de la patologías autoinmunes. El SPG tipo 2 se inicia en la vida adulta y no es atingente a nuestro caso.
Entre las patologías congénitas que cursan con destrucción de las glándulas suprarrenales se encuentra la Adrenoleucodistrofia ligada al cromosoma X (ALD-X), la Hiperplasia congénita lipoídea (Deficiencia Star), y los déficit de p-450 óxido reductasa o de lipasa ácida (Enfermedad de Wolman).
La ALD-X es una enfermedad progresiva que se asocia a desmielinización del sistema nervioso central y periférico, insuficiencia suprarrenal, compromiso testicular5 y acumulación de ácidos grasos de cadena muy larga (AGCML > 22 carbonos). Se hereda de modo recesivo ligada al cromosoma X, y es causado por la mutación del gen ABCD1, localizado en Xq286 que codifica para la proteína ALD-P, perteneciente al grupo de transportadores ABC (ATP-binding cassette) del peroxisoma. El trastorno bioquímico asociado a este defecto se traduce en la incapacidad del peroxisoma para degradar ácidos grasos saturados de cadena muy larga, produciendo así su acumulación en el plasma, y causando las manifestaciones clínicas en los pacientes afectados6.
Presentamos el caso de un niño que debuta con estrabismo unilateral y enfermedad de Addison, hechos que unidos a las alteraciones características de sustancia blanca detectadas en la RN cerebral, permitieron identificar su etiología.
Caso clínico
Niño de 7 años de edad, segundo hijo de padres no consanguíneos, sin antecedentes de patología perinatal, con historia de crecimiento y desarrollo normal. Consulta por estrabismo de ojo izquierdo de un mes de evolución. Su madre refiere que presenta astenia y adinamia vespertinas, sin avidez por el agua y la sal.
Al examen físico, lo destacable era un paciente con poca conexión con el medio, con labilidad emocional, normotenso, con sobrepeso (p81; 1,4 DS), talla 130 cm (p93; 1,51 DS), prepuber, con pigmentación de las encías y cicatrices cutáneas, exotropia de ojo izquierdo y conservación de los movimientos oculares.
La atención al medio y el rendimiento escolar había disminuido en los últimos meses.
Debido a la rápida progresión de su estrabismo, se solicita RN cerebral, que muestra leucoencefalopatía con marcada alteración de la sustancia blanca periventricular a nivel atrial, que se extiende a los cuerpos geniculados laterales, a las radiaciones ópticas, alrededor de los cuernos occipitales y al esplenio del cuerpo calloso con compromiso de los tractos córtico espinales desde el mesencéfalo hasta la protuberancia (Figura 1).
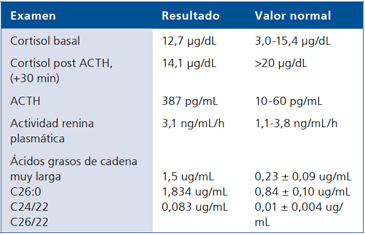
Por la historia de astenia y adinamia, asociada a pigmentación de encías y cicatrices de la piel, se planteó una insuficiencia suprarrenal, confirmada con los exámenes de laboratorio (Tabla 1); ello, en conjunto con las alteraciones en las neuroimagenes, sustentan la sospecha de Adrenoleucodistrofia ligada al X (ALD-X). La medición de ácidos grasos de cadena muy larga en plasma (Tabla 1) señaló niveles elevados de C26:0 y de la relación C24/22, C26/22, perfil característico y confirmatorio de ALD-X.
Se inició tratamiento con Cortisol (10 mg/m2/d) y Florinef ® (0,05mg/d).
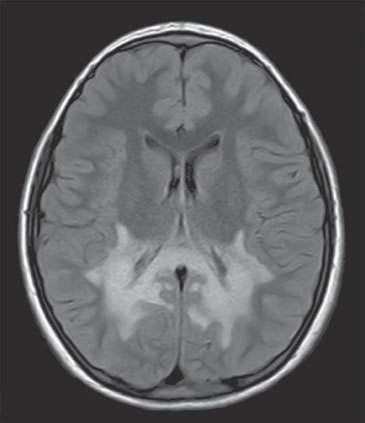
Figura 1. Resonancia Nuclear cerebral. Se observa una leucoencefalopatía con marcada alteración de la sustancia blanca periventricular, que se extiende a los cuerpos geniculados laterales y radiaciones ópticas alrededor de los cuernos occipitales.
Tabla 1. Exámenes realizados al paciente
Discusión
Presentamos el caso de un niño con enfermedad de Addison, asociada a leucoencefalopatía y aumento de la concentración plasmática de ácidos grasos de cadena muy larga, conjunción que sustenta el diagnóstico de ALD-X. El daño suprarrenal por inclusiones lipídicas genera una merma en la producción de cortisol, lo cual estimula la liberación hipotalámica de CRH, con el consiguiente aumento de la secreción hipofisiaria de ACTH2.
La ALD es una enfermedad que pertenece al grupo de las leucoencefalopatías y se hereda ligada al cromosoma X. Tiene una prevalencia entre 1:20.000 y 1:50.000 hombres nacidos vivos, sin preferencia geográfica ni racial, siendo el desorden peroxisomal hereditario más común. Se han descrito tres fenotipos principales: la forma cerebral infantil, la adrenomieloneuropatía (AMN) y el de la enfermedad de Addison aislada6.
El fenotipo cerebral infantil, como lo es nuestro paciente, se inicia entre los 4 y 8 años de edad, con síntomas sugerentes de déficit atencional, que evoluciona hacia el compromiso cognitivo, conductual, auditivo, visual y motor, llegando a una discapacidad total en un período variable de tiempo, que generalmente no sobrepasa 2 años. En la AMN, predomina la desmielinización de la médula espinal y del sistema nervioso periférico; se presenta alrededor de los 20 a 30 años de edad, con paraparesia progresiva, alteración del control de esfínteres, disfunción suprarrenal y sexual. Este fenotipo, en alrededor del 20% de los casos se presenta en mujeres, en etapas más tardías de la vida. El fenotipo de enfermedad de Addison aislada, se manifiesta a partir de los 7 años de edad, sin signos de compromiso de SNC, aunque algunos pueden tener manifestaciones de AMN. Los pacientes pre-sintomáticos pueden ser diagnosticados debido al estudio de un familiar afectado. Las forma con compromiso del sistema nervioso central es más rápidamente progresiva que la AMN7,8.
La fisiopatología no es conocida a cabalidad, pero se sabe que la mutación en el gen ABDC1 altera la funcionalidad de la proteína ADP, encargada de transportar los AGCML al interior del peroxisoma para su degradación. Como consecuencia los ácidos grasos se acumulan en el plasma y en los tejidos. De ellos, los que producen mayor daño oxidativo son el C26 y el C24, el cual es tanto directo como indirecto; inducen además liberación de citoquinas que llevan a la activación inmune, la cual es responsable de la degradación de la mielina. Esto causa degeneración progresiva del sistema nervioso central y atrofia final de las suprarrenales y los testículos, originando insuficiencia de ambas funciones glandulares.
La microscopía electrónica ha evidenciado inclusiones citoplasmáticas lamelares, que contienen AGCML, en las glándulas suprarrenales, en las células de Schwan y de Leydig. Con el avance de la enfermedad, se produce atrofia suprarrenal.
La ISRP se presenta en el 90% de los pacientes con fenotipo cerebral infantil y en el 70% de los que tienen AMN9. La edad más común de presentación es alrededor de los 7 años, pero puede ocurrir entre los 2 años y la adultez10. Korenke y cols5, describieron disfunciones endocrinológicas en 55 pacientes con ALD-X, y encontraron falta de correlación entre el genotipo y el trastorno endocrinológico presentado.En el 8% de los casos la insuficiencia suprarrenal es la única expresión clínica de ALD-X. El compromiso endocrinológico es más común en niños y adolescentes, es decir en las formas de manifestación temprana de la enfermedad. En la serie señalada la elevación de ACTH, y el aumento de la actividad de renina plasmática se explican por la deficiencia de mineralocorticoides; ellos son los exámenes más frecuentemente alterados. Como dato importante resaltó la calvicie como un hallazgo constante tanto en ALD como AMN11-13.
El tratamiento de la enfermedad de Addison no difiere del propio de otras formas de insuficiencia suprarrenal, y consiste en el reemplazo fisiológico de glucocorticoides (cortisol 8-12 mg/m2/d) y mineralocorticoides (FlorinefR 0,05-0,1 mg/d), con lo cual se controla la pérdida de sodio y se normaliza el potasio.
Más complejo es el tratamiento de la enfermedad de base ya que la ALD-X, no tiene a la fecha tratamiento curativo. En las décadas de 1970 y 1980 se trató a los pacientes con aceite de Lorenzo (20% ácido erúcico C22:1 y 80% ácido oleico C18:1) pero se demostró que aunque disminuían los niveles de AGCML, no influía en el curso de la enfermedad14.
Este compuesto no traspasa la barrera hematoencefálica, quedando así sin acción sobre el SNC y tampoco constituye un aporte significativo en la actividad de enzimas antioxidantes; su uso actual es discutible.
Se ha intentado también, aunque con poco éxito, el transplante de médula ósea. Hay descritas respuestas favorables en niños y adolescentes con evidencia precoz de fenotipo neurológico. Se especula que podría generar impacto positivo sobre la progresión de la enfermedad, pues las células trasplantadas podrían degradar AGCML en el SNC15. La indicación actual está focalizada a pacientes con RMN cerebral alterada, sin déficit neurológico o en estado temprano de la enfermedad. Sin embargo, en un metaanálisis de 372 pacientes con este diagnóstico, Moser y cols16, consigna el transplante de médula ósea en 59 de ellos; de éstos el 34% falleció durante el primer año, y el 60% de los sobrevivientes mantuvo el nivel de su enfermedad o empeoró.
En estos pacientes la muerte ocurrió por progresión de la enfermedad o por complicaciones propias del procedimiento (transplante), el cual en sí de alto riesgo. No es este un procedimiento recomendado en pacientes en estado avanzado de la enfermedad, pues podría acelerar su progresión. Tampoco está recomendado en AMN, pues en ellos predomina el compromiso periférico, ni tampoco en pacientes con RMN normal16.
La terapia de reemplazo con gluco y mineralocorticoides es efectiva en corregir la insuficiencia suprarrenal asociada con la ALD-X, pero no influye sobre el pronóstico neurológico de la enfermedad17.
En conclusión, dado que la ALD-X afecta en forma importante y frecuente a las glándulas suprarrenales se recomienda buscar dirigidamente este compromiso en niños o adultos con ISRP. Del mismo modo, es recomendable también, investigar ISRP en todo paciente con diagnóstico de ALD-X. Un diagnóstico precoz permite adoptar decisiones terapéuticas que influyen en el curso de la enfermedad.
Referencias
- Arlt W, Allolio B 2003. Adrenal insufficiency. Lancet 361: 1881- 1893.
- Martínez A, Lizama M, Reyes ML, Cattani A. 2007. Insuficiencia suprarrenal primaria de etiología autoinmune: dos casos clínicos. Rev Chil Pediatr 78 (3): 292-300.
- Ten S, New M, Maclaren N. 2001. Clinical Review 130. Addison disease 2001. J Clin Endocrinol Metab 86 (7): 2909-2921.
- Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. 2005. Primary adrenal insufficiency in children: twenty years experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab 90: 3243-3250.
- Korenke C, Roth C, Krasemann1 E, Hüfner M, Hunneman DH, Hanefeld F. 1997. Variability of endocrinological dysfunction in 55 patients with X-linked adrenoleucodystrophy: clinical, laboratory and genetic findings. Eur J Endocrinol 137: 40-47.
- Moser A, Steinberg S, Raymond G. 2009. X-Linked Adrenoleukodystrophy. Disponible en: www.genetest.org. consultado en June 2, 2009.
- Melhem E, Barker P, Raymond G, Moser H. 1999. X-Linked Adrenoleukodystrophy in Children: Review of Genetic, Clinical, and MR Imaging Characteristics. AJR 173: 1575-1581.
- Kim J H, Kim H J. 2005 Childhood X-linked Adrenoleukodystrophy: Clinical-Pathologic Overview and MR Imaging Manifestations at Initial Evaluation and Follow-up. Radio Graphics 25: 619-631.
- Fourcade F, López Erauskin J, Galino J, Duval C, Naudi A, Jove M, et al. 2008. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet 17 (12): 1762-1773.
- García Cuartero B, González Vergaz A, Herranz Antolín S, Blanco C, Sánchez Mateos M, Carrasco Marina L, et al. 2008. Adrenoleucodistrofia ligada al cromosoma X: diagnóstico olvidado en niños con enfermedad de Addison idiopática. An Pediatr (Barc) 68 (4): 401-414.
- Ronghe MD, Barton J, Jardine PE, Crowne EC, Webster MH, Armitage M, et al. 2002. The importance of testing for adrenoleucodystrophy in males with idiopathic Addiso's disease. Arch Dis Child 86: 185-189.
- Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. 2005. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr 146: 528-532.
- Laureti S, Casucci G, Santeusanio F, Angeletti G, Aubourg P, Brunetti P. 1996. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addison's disease in young adult patients. J Clin Endocrinol Metab 81 (12): 470-474.
- Deon M, Wajner M, Sirtori LR, Fitarelli D, Coelho DM, Sitta A, et al. 2006 The effect of Lorenzo's oil on oxidative stress in X-linked adrenoleukodystrophy. J Neurol Sci 247: 157-164.
- Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. 2005. Primary Adrenal Insufficiency in Children: Twenty Years Experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab 90 (6): 3243-3250.
- Moser HW, Loes DJ, Melhem ER, Raymond GV, Bezman L, Cox CS, et al. 2000. X-Linked Adrenoleukodystrophy: Overview and Prognosis as a Function of Age and Brain Magnetic Resonance Imaging Abnormality. A Study Involving 372 Patients. Neuropediatrics 31: 227-239.
- Moser HW, Smith KD, Watkins PA, Powers J, Moser AB. X-linked Adrenoleukodystrophy. En: Valle, Beaudet, Vogelstein, Kinzler, Antonarakis, Ballabio Editores. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill. Disponible en www.ommbid.com