Paresia del III par craneal como manifestación de un corticotropinoma silente
Sara Gómez R.1, Isabel Pavón de P.1, Guadalupe Guijarro de A.1, Clara Euduardina Torán R.1, Naiara Modroño M.1, Joanny Duarte L.2 y Juan Carlos Gómez A.3
Third nerve palsy as an expression of a corticotroph adenoma
1Servicio de Endocrinología y Nutrición del Hospital
Universitario de Getafe.
2Servicio de Anatomía Patológica del Hospital
Universitario de Getafe.
3Servicio de Neurocirugía del Hospital Universitario de
Getafe.
Conflicto de intereses: Ninguno de los autores presenta
relación financiera o personal que pudiera dar lugar a un
conflicto de intereses.
Correspondencia a:
Sara Gómez Rodríguez
Hospital Universitario de Getafe.
Carretera de Toledo Km 12,500. CP: 28905-Getafe
Teléfono: 91-683 93 60 Fax: 91-683974.
E mail: patsara8@hotmail.com / sgomezr88@gmail.com
Recibido: 13-02-2015
Aceptado: 23-05-2015
Silent corticotroph adenomas (SCA) are pituitary adenomas inmunoreactive to ACTH without hormonal overproduction and they form the 3% of the pituitary adenomas. Unlike the ademonas that cause the Cushing’s disease, frequently the SCA are aggressive macroademonas and their clinical expressions are the compromise of nearby structures with or without pituitary apoplexy. Frequently, the treatment chosen is a surgical procedure, albeit the total resection difficulties. The SCA are not diagnosed until an anatomopathological and immunohistochemical study is carried out. In this document, a case study of a woman diagnosed with a clinically silent macrocorticotroph adenoma discovered by a third nerve palsy is explained.
Key words: Corticotroph adenoma,silent adenoma, pituitary.
Mujer de 59 años sin antecedentes de interés que consultó por mareo y dificultad para la movilización del ojo derecho objetivándose en la exploración física una paresia del III par craneal derecho sin afectación pupilar. Por este motivo se realizó un TAC craneal urgente que mostró una masa de localización intra y supraselar que remodelaba la silla turca, con extensión a seno esfenoidal derecho, afectando el seno cavernoso ipsilateral y con porción superior de apariencia quística, valorándose como primera opción diagnóstica un macroadenoma hipofisario.
Se realizó una campimetría que evidenció una hemianopsia heterónima temporal. Una RM (Resonancia Magnética) posterior confirmó el hallazgo de adenoma hipofisario de 42 x 34 x 25 mm que realzaba moderadamente con en el contraste paramagnético, con varios quistes de retención glandular (el mayor de aproximadamente 1 cm de diámetro), con extensión supraselar e invasión del seno esfenoidal y seno cavernoso derecho con desplazamiento y compresión del quiasma óptico y del tallo hipofisario (Figuras 1 y 2).
Interrogada específicamente refería cefalea retrocular y negaba síntomas de déficit y de sobreproducción hormonal.
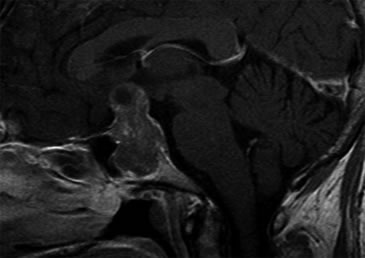
Figura 1. Corte sagital en secuencia T1 con gadolinio.
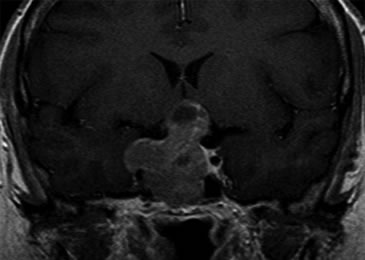
Figura 2. Corte coronal en secuencia T1 con gadolinio.
En la exploración física presentaba limitación para mover el globo ocular hacia arriba, abajo y adentro, con función pupilar normal, todo ello compatible con una paresia o parálisis incompleta del III par de ojo derecho. No se objetivó otros signos relevantes.
El estudio hormonal reveló un hipotiroidismo central: T4-L: 0,7 ng/dL [0,90-1,70], con TSH: 0,37 μU/ml [0,40-4,78] y T3-L: 1,28 pg/mL [2-4,40]; cifra de prolactina 41,6 ng/ml [4,80-23,30] e hipogonadismo hipogonadotropo: FSH: 1,9 mUI/mL [4-33,4], LH < 0,1 mUI/mL [0,5-73,3], Estradiol: 5 pg/mL [19,5-356,7]. No se pudo valorar el eje hipotálamo-hipofiso-adrenal por haberse iniciado previamente tratamiento con Dexametasona. Se inició tratamiento con levotiroxina.
La paciente fue intervenida por vía transesfenoidal no siendo posible la extirpación completa del tumor. Como complicaciones se produjo una fístula del LCR, que precisó la colocación de un drenaje lumbar. Tras la cirugía, la afectación del III par craneal revirtió.
El diagnóstico AP fue de adenoma hipofisario con células muy uniformes, núcleos pequeños y moderada cantidad de citoplasma eosinófilo. No se observaba atipia significativa ni actividad mitósica. Con técnica de reticulina se observaba pérdida de patrón hipofisario habitual. El estudio inmunohistoquímico mostraba intensa positividad para ACTH, positividad de células aisladas para prolaptina, negatividad para GH, LH, TSH y FSH. El Ki 67 fue positivo para menos del 1% (Figura 3).
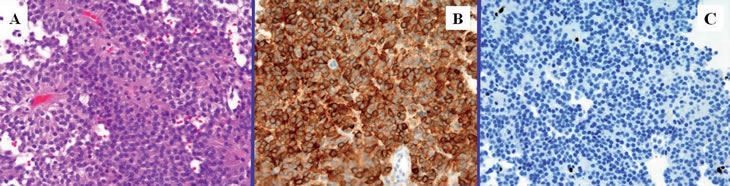
Figura 3. Estudio AP. A) Proliferación de hábito organoide, con crecimiento sólido en torno a una rica vascularización capilar; B) ACTH positivo; C) Ki-67 débilmente positivo (inferior al 1%).
Tras la cirugía se objetivaron restos tumorales que permanecieron
estables durante 2 años con persistencia del
déficit de TSH. Tras el cese del tratamiento corticoideo
perioperatorio se descartó hipercortisolismo. Dos años
después de la cirugía se evidencia en nueva RM aumento
leve del tamaño de los restos tumorales, manteniéndose
asintomática en todo momento. Se plantea radioterapia vs
nueva cirugía.
Antes de iniciarse un nuevo tratamiento, la paciente acudió al Servicio de Urgencias por clínica compatible con paresia del III par derecho y cefalea periorbitaria. Fue reintervenida quirúrgicamente con resección aparentemente completa de la tumoración.
El estudio AP fue similar al inicial. El estudio funcional tras la segunda cirugía mostró persistencia del déficit de TSH y gonadotropinas, estando la paciente correctamente sustituida con 75 mcg de levotiroxina.
Tras esta segunda cirugía y tras comprobar la persistencia de restos, se remitió a la paciente para tratamiento radioterápico.
Recibió radioterapia estereoatáxica fraccionada (50,4 Gy en seis semanas. Dosis fraccionada de 1,8 Gy). Desde entonces se mantiene estable, sin evidencia de progresión de restos y sin nuevos datos de hipopituitari.
Discusión
Los tumores hipofisarios constituyen el 15% de las neoplasias intracraneales y se descubren en 3-27% de las necropsias como hallazgo incidental1-7.
Aproximadamente el 15% de los adenomas hipofisarios son inmunoreactivos a ACTH, el 10% cursan con hipercortisolismo y el 3% mantienen una secreción normal de cortisol1, constituyendo un subtipo de adenomas hipofisarios conocidos como corticotropinomas silentes, la mayor parte macroadenomas al diagnóstico.
Al igual que en los otros macroadenomas no funcionantes (ANF), la ausencia de clínica hormonal ocasiona retraso en el diagnóstico, hasta que el hallazgo incidental o la clínica por compromiso de espacio hace que se descubran estos tumores. Las manifestaciones clínicas son similares siendo más frecuentes las alteraciones visuales 67-86,7%8-10 que pueden deberse a compresión del quiasma óptico, infiltración directa de los nervios ópticos o hidrocefalia por obstrucción del flujo del LCR11. La manifestación más típica es la hemianopsia bitemporal que nuestra paciente también presentaba.
La afectación de los oculomotores aparecen en el 13%10 secundario a la compresión del seno cavernoso y su contenido nervioso, causando parálisis de los pares craneales III, IV y VI. La clínica de debut de nuestra paciente fue paresia del III par.
La cefalea se presenta en el 37-70% de los casos8-10 sin que exista correlación entre el tamaño tumoral y su intensidad. Nuestra paciente no refería espontáneamente cefalea, sólo en el interrogatorio dirigido contaba cefalea retrocular.
La RM es la técnica de elección para el estudio de imagen con la que se demuestra ocupación de la hipófisis. El hallazgo de un quiste en su interior puede interpretarse como inespecífico, sin embargo, Cazabat et al proponen este hallazgo como predictor de la estirpe celular. Observaron quistes hiperintensos en T2 en el 100% de los CS (en su mayoría múltiples microquistes de < 3 mm), en el 50% de los corticotropinomas funcionantes y en el 17% de los adenomas no funcionales por lo que postulan que el descubrimiento de múltiples microquistes tiene una especificidad de casi el 95% para CS12. En nuestro caso, la lesión mostraba varios quistes en su interior.
La ACTH puede ser normal o levemente elevada y no se acompaña de hipercortisolismo secundario. Este fenómeno puede ser debido a un aumento del catabolismo lisosómico, a la existencia de un complejo de Golgi menos desarrollado que condicionaría un almacenamiento inadecuado de corticotropina en los gránulos neurosecretores y a la secreción de hormona biológicamente inactiva. Los CS generan en su mayoría, una ACTH disfuncional debido a una menor expresión de propiomelanocortina por una alteración en su factor de transcripción principal, T pit. Como resultado, se sintetiza una molécula de mayor peso molecular (big ACTH) que tiene una actividad biológica escasa. Esta molécula se puede detectar en cromatografía de filtración en gel13,14. La interrupción del acceso de las hormonas hipotalámicas y la dopamina a la hipófisis por “compromiso del tallo”, se traduce en hiperprolactinemia con pérdida simultánea de otras hormonas hipofisarias: hipopituitarismo 26-11%8-10 e hipogonadismo central 17%8. En nuestro caso se evidenció una hiperprolactinemia leve compatible con compresión del tallo hipofisario y un déficit de gonadotropinas y TSH.
Debido a la ausencia de clínica propia, el descubrimiento de la estirpe celular mediante el estudio inmuninohistoquímico de la pieza quirúrgica es fundamental para el diagnóstico. La mayor parte de los casos corresponden a adenomas densamente granulados, tintorialmente basófilos. Presentan grados variables de positividad para ACTH, y pueden expresar otros componentes de POMC15. El anticuerpo Ki-67 permite estimar la actividad proliferativa y determinar si existen diferencias según el fenotipo morfológico16. Los CS pueden ser morfológicamente indistinguibles de los adenomas corticotropos, o bien pueden presentar características atípicas como mayor grado de pleomorfismo, actividad mitótica y mayor actividad proliferativa con Ki-6717. En nuestro caso, presentaba un Ki-67 positivo en menos del 1% y poca actividad mitótica.
La planificación terapéutica de un adenoma hipofisario silente debe realizarse en relación a su tamaño. Cuando se trata de un microadenoma (< 1 cm), la opción elegida es la observación. Si por el contrario, nos encontramos ante un macroadenoma, como en la mayoría de los CS, nos inclinaremos hacia el intervencionismo. La cirugía transesfenoidal es el tratamiento de elección.
En la mayoría de las series se ha descrito un comportamiento agresivo de los CS con tendencia al crecimiento y a la recidiva, como sucedió en nuestro caso. Cooper et al encuentran una tasa de recurrencia del 63%, 5 años antes que en los ANF (3 vs 8 años)8.
La mayor agresividad e invasión de estructuras como el seno cavernoso, traduce una vía biológica diferente de los ANF Los marcadores de crecimiento tumoral como el Ki-67 o el MIB-1 son empleados para establecer el pronóstico de los CS. El desarrollo de hipercortisolismo es un indicador de agresividad y futura malignidad y aparece sobre todo en las recidivas. Este hecho puede deberse a una posterior síntesis de ACTH biológicamente activa y a una necesidad de niveles más bajos de esta hormona para producir enfermedad de Cushing14,18,19.
El tratamiento médico con análogos de somatostatina y los agonistas dopaminérgicos se ha utilizado tras cirugía, observándose disminución del tamaño del adenoma en algunas series aunque los resultados son muy poco esperanzadores20.
La radioterapia está indicada para tratar y prevenir las recurrencias tras la cirugía, como se realizó en nuestro caso. Se observa recidiva en el 42% de la población que no recibe radioterapia postquirúrgica frente al 26% que sí lo hace9. Asimismo, se ha utilizado para reducir el tamaño de restos postquirúrgicos. Varias series han ido encaminadas a la demostración de la efectividad y la menos agresividad de la radioterapia como tratamiento primario. Kajiwara et al explican que los diferentes tipos de radioterapia (estereotáctica, GammaKnife, CyberKnife o LINAC system) son efectivos en los adenomas no funcionales ya que lograron controlar el tumor en más del 90% de los casos21.
Debido a la ausencia de hipercortisolismo, el retraso en el diagnóstico es frecuente, encontrándonos en la mayoría de los casos, macroadenomas con el consiguiente compromiso de estructuras vecinas que dificultan la técnica quirúrgica. Además, constituyen un porcentaje elevado dentro de los adenomas hipofisarios y con frecuencia presentan un curso agresivo y recurrente. Por lo que, en conclusión, el conocimiento de los CS es necesario para un correcto manejo terapéutico precoz.
Referencias bibliográficas
- Melmed S. 2003. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 11: 1603-1618.
- Teramoto A, Hirakawa K, Sanno N, Osamura Y. 1994. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 1: 161-164. [PubMed].
- Parent AD, Brown B, Smith EE. 1982. Incidental pituitary adenomas: a retrospective study. Surgery 5: 880-883.
- Kontogeorgos G, Kovacs K, Horvath E, Scheithauer BW. 1991. Multiple adenomas of the human pituitary. A retrospective autopsy study with clinical implications. J Neurosurg 2: 243-247.
- Tomita T, Gates E. 1999. Pituitary adenomas and granular cell tumors. Incidence, cell type, and location of tumor in 100 pitu itary glands at autopsy. Am J Clin Pathol 6: 817-825.
- Molitch ME, Russell EJ. 1990. The pituitary “incidentaloma”. Ann Intern Med 12: 925-931.
- Buurman H, Saeger W. 2006. Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol 5: 753-758.
- Cooper O, Ben-Shlomo A, Bonert V, Bannykh S, Mirocha J, Melmed S. 2010. Silent corticogonadotroph adenomas: clinical and cellular characteristics and long-term outcomes. Horm Cancer 2: 80-92.
- Webb KM, Laurent JJ, Okonkwo DO, Lopes MB, Vance ML, Laws ER. 2003. Jr Clinical characteristics of silent corticotro phic adenomas and creation of an internet-accessible database to facilitate their multi-institutional study. Neurosurgery 5: 1076-1084. discussion 1084-1085.
- Scheithauer BW, Jaap AJ, Horvath E, Kovacs K, Lloyd RV, Meyer FB, Young WF. 2000. Clinically silent corticotroph tumors of the pituitary gland. Neurosurgery 3: 723-729. discussion 729-730.
- Martínez Quiñones JV, de la Torre Gutiérrez Ml, Serrano Martín P. 1999. Manifestaciones poco frecuentes de la hidrocefalia. A propósito de un caso. Neurocirugía 10: 475-476.
- Cazabat L, Dupuy M, Boulin A, Bernier M, Baussart B,
Foubert L, et al. 2014. Silent, but not unseen: Multi-microcystic
aspect on T2-weighted MRI in Silent Corticotroph Adenomas. Clin Endocrinol (Oxf). - Aznar Rodríguez SM, Moreno Pérez O, Revert Marrahí P, Martínez Fuster S, Boix Carreño S, Picó Alfonso AM. 2008. Corticotropinomas silentes: a propósito de dos casos. Endocrino l Nutr 55 (8): 367-372.
- Kageyama K, Oki Y, Nigawara T, Suda T, Daimon M. 2014. Pathophysiology and treatment of subclinical Cushing´s disease and pituitary silent corticotroph adenomas. Endocrine Journal 1 0: 941-948.
- Niveiro de Jaime M, Aranda López FI, Peiró Cabrera G. 2003. Patología de los adenomas hipofisarios. Rev Esp Patol 36 (4): 257-372.
- Knosp E, Kitz K, Perneczky A. 1989. Proliferation activity in pituitary adenomas measurement by monoclonal antibody ki-67. Neurosurgery 25: 927-930.
- Roncaroli F, Scheithauer BW, Young WF, Horvath E, Kovacs K, Kros JM, et al. 2003. Silent corticotroph carcinoma of the adenohypophysis: a report of five cases. Am J Surg Pathol 27: 477-486.
- Baldeweg SE, Pollock JR, Powell M, Ahlquist J. 2005. A spectrum of behaviour in silent corticotrop pituitary adenomas. Brithish Journal of Neurosurgery 19 (1): 38-42.
- Alahmadi H, Lee D, Wilson JR, Hayhurst C, Mete O, Gentili F, et al. 2012. Clinical features of silente corticotroph adenomas. Acta Neurochir 154: 1493-1498.
- Mayson SE, Snyder PJ. 2014. Silent (clinically nonfunctioning) pituitary adenomas. J Neurooncol 117 (3): 429-436.
- Kajiwara K, Saito K, Yoshikawa K, Ideguchi M, Nomura S, Fujii M, et al. 2010. Stereotactic radiosurgery/radiotherapy for pituitary adenomas: a review of recent literature. Neurol Med Chir (Tokyo) 50 (9): 749-755.
- Gilsanz A, Moreno B, Obiols G, Zugasti A, Catalá M, Lucas T, et al. 2006. Guía clínica del diagnóstico y tratamiento de los tumores hipofisarios no funcionantes y gonadotropinomas. Endocrinol Nutr 53 (01): 13-18.