Hipoglicemia infantil
Vivian Gallardo T.1,2 y Verónica Mericq G.1,3
Hypoglycemia of infancy
1Instituto de Investigaciones Materno Infantil, Universidad
de Chile, Hospital San Borja Arriarán.
2Becada Endocrinología Infantil y Diabetes.
3Endocrinóloga Infantil, Clínica Las Condes.
Correspondencia:
Verónica Mericq G.
Santa Rosa 1234, Hospital San Borja Arriarán, IDIMI
E-mail: vmericq@med.uchile.cl
Recibido: 27 de Agosto de 2010
Aceptado: 7 de Septiembre de 2010
Hypoglycemia of infancy is a common metabolic disorder that can have serious neurological consequences. Therefore, its early diagnosis and treatment are crucial prognostic factors. Hypoglycemia has a variety of causes and a good clinical history, physical examination and laboratory determination will orient the correct diagnosis. Occasionally a molecular study will be required.
Key words: Transient hypoglycemia, persistent hypoglycemia, hypoglycemia, childhood and adolescence, causes, diagnosis and therapy hypoglycemia.
Definición
La hipoglicemia constituye una de las alteraciones metabólicas más importantes durante la niñez cuyo diagnóstico y tratamiento precoz son fundamentales en la prevención de secuelas neurológicas1.
La definición de hipoglicemia ha sido controversial; décadas atrás se utilizaron puntos de corte según la edad gestacional y las horas de vida2. Hoy en día, se considera hipoglicemia una concentración de glicemia < 50 mg/dL a cualquier edad, ya que a ese nivel se producen cambios homeostáticos en los sistemas nervioso central y hormonal; con valores por debajo de 50 mg/dL se han evidenciado anomalías somatosensoriales3.
Fisiología de la regulación de la glicemia
La glucosa es la principal fuente de energía para el cuerpo humano y todas las células del organismo la necesitan para mantener sus funciones vitales. El principal aporte proviene de los carbohidratos de la dieta; durante el período postprandial, al disminuir su nivel, se produce la degradación del glicógeno hepático (glicogenolisis) proceso bajo el control de catecolaminas y glucagón. Al depletarse estos depósitos (4-6 horas en recién nacidos, 6-8 horas en lactantes y 14-20 horas en el adulto), se inicia la neoglucogénesis a partir de sustratos periféricos como alanina, lactato y glicerol, provenientes del músculo, del metabolismo anaeróbico y de los depósitos de grasa, respectivamente; este proceso es estimulado por cortisol y hormona de crecimiento e inhibido por insulina. Además, se estimula la cetogénesis a partir de la b-oxidación de ácidos grasos, que sirven como fuente de energía alternativa a la glucosa, especialmente para el sistema nervioso central. Este proceso es estimulado por glucagón e inhibido por insulina.
Cualquier circunstancia que disminuya la producción, aumente la utilización de glucosa o ambas posibilidades, puede desencadenar hipoglicemia.
Síntomas
Neuroglucopénicos y autonómicos. En el RN y lactantes, los síntomas de hipoglicemia son inespecíficos, tales como irritabilidad, letargia, cianosis, hipotonía, dificultad para alimentarse, taquipnea, apnea, bradicardia, hipotermia, los que se pueden confundir con muchas otras patologías. En niños mayores los síntomas iniciales causados por respuesta autonómica son sudoración, debilidad, taquicardia, temblores, sensación de hambre, seguidos de síntomas neuroglucopénicos como letargia, irritabilidad, confusión, cambios de comportamiento (que pueden confundirse con enfermedad psiquiátrica) hasta convulsiones y coma.
Tratamiento inicial
Cuando se sospecha hipoglicemia, ella debe ser confirmada con una glicemia capilar, siendo necesario obtener una muestra de sangre antes de la administración de glucosa para análisis posterior; esta es la llamada “muestra crítica” que nos orientará en el diagnóstico. También se debe tomar concomitantemente una muestra de orina. Una vez conseguida esta muestra, si el paciente está conciente, se realiza una prueba con glucagón im 0,1 mg/kg (máximo 1 mg) tomando una glicemia 30 minutos después; un aumento > 30 mg/dL respecto al basal es una respuesta positiva e indica que los depósitos de glicógeno hepático no estaban depletados en el momento de la hipoglicemia siendo altamente sugerente de hiperinsulinismo. Posteriormente, si el paciente es capaz de ingerir por boca se debe dar un carbohidrato de rápida absorción; si la glicemia no mejora en 10-15 minutos se debe administrar glucosa parenteral. Si el paciente está inconsciente o es incapaz de ingerir por boca, debe ser tratado con glucosa intravenosa, con un bolo inicial de solución glucosada al 10%, 2 cc por kg administrado en 2-3 minutos, seguido de una infusión continua de glucosa para mantener glicemia en márgenes seguros (entre 70-120 mg/dL).
Consideraciones a tomar en cuenta en el análisis de los valores de glicemia
La glicemia capilar es muy variable en el área de la hipoglicemia por lo que siempre se debe confirmar con un método de laboratorio (glucosa oxidasa o glucosa-6-fosfato deshidrogenasa). Los valores de glucosa en sangre total son aproximadamente 15% menores a los del plasma y pueden ser aún menores si el hematocrito es alto. El procesamiento de la muestra debe hacerse a la brevedad, ya que los valores de glucosa caen 15-20 mg/dL por cada hora que la muestra permanezca a temperatura ambiente. Para el análisis hormonal la muestra debe ser centrifugada y el plasma congelado a -20º C.
En hipoglicemia espontánea la muestra crítica es fundamental para la orientación de su causa y evitar exponer al niño a una nueva hipoglicemia inducida con una prueba de ayuno. En la Tabla 1 se detallan los exámenes a solicitar en la muestra crítica.
Una respuesta normal a la hipoglicemia corresponde al aumento de cuerpos cetónicos y ácidos grasos libres, disminución de lactato, con insulina indetectable pero con aumento de las otras hormonas de contrarregulación. En la Figura 1 se muestra un algoritmo de análisis de la muestra crítica.
Clasificación
La hipoglicemia se clasifica como transitoria si ocurre
dentro de las primeras 48 horas de vida y persistente si ocurre
y/o persiste más allá de las 48 horas de vida.
Tabla 1. Muestra crítica

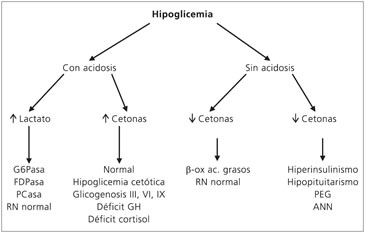 Figura 1. Interpretación de la
muestra crítica.
Abreviaciones: G6Pasa: glucosa-
6-fosfatasa; FDPasa: fructosa-
1,6-difosfatasa; PCasa: piruvato
carboxilasa; PEG: pequeño
para la edad gestacional; ANN:
asfixia neonatal
Figura 1. Interpretación de la
muestra crítica.
Abreviaciones: G6Pasa: glucosa-
6-fosfatasa; FDPasa: fructosa-
1,6-difosfatasa; PCasa: piruvato
carboxilasa; PEG: pequeño
para la edad gestacional; ANN:
asfixia neonatal
Causas de hipoglicemia transitoria
- Prematurez, por disminución de las reserva de glicógeno hepático, ya que éste se deposita en las últimas semanas de gestación. También existe inmadurez de las enzimas gluconeogénicas y cetogénicas.
- Pequeño para la edad gestacional por disminución de las reservas de glicógeno debido a las bajas concentraciones de insulina intrauterina e hipoxia crónica que produce ineficiente utilización anaeróbica de la glucosa.
- Hijo de madre diabética: La hiperglicemia materna causa hiperglicemia fetal que estimula al páncreas fetal a producir insulina (favorece la macrosomía); al caer el flujo útero-placentario se produce un descenso brusco de la glicemia en presencia de niveles de insulina elevados, generando hipoglicemia.
- RN con asfixia o sepsis: En situaciones de pobre oxigenación o disminución de la perfusión de tejidos periféricos la energía se obtiene de la glicólisis anaeróbica que es aproximadamente 5% de la producida por glicólisis aeróbica, incrementando así la utilización de glucosa y produciendo hipoglicemia4.
- Otras: La eritroblastosis fetal por aumento del consumo, y los catéteres umbilicales altos cercanos a la mesentérica superior y la interrupción de perfusión con suero glucosado producen en forma rápida hipoglicemia por un mecanismo similar al anterior.
Causas de hipoglicemia persistente
El análisis por grupo etario permite una orientación diagnóstica más eficiente; en el RN las tres causas más frecuentes son: hiperinsulinismo, déficit hormonales, déficit enzimáticos. En el lactante mayor de 1 año la primera causa es la hipoglicemia cetósica y en el niño mayor las intoxicaciones e insulinomas.
I. Trastornos del metabolismo
Alteración del metabolismo de los hidratos de carbono
- Alteración de la glucogenólisis: A continuación se describen los defectos más frecuentes, todos ellos con herencia de tipo autosómica recesiva.
- Deficiencia de glicógeno sintetasa (Glucogenosis tipo 0), enzima que cataliza la formación de enlaces 1-4 de glucosa para formar glicógeno. Su déficit produce hipoglicemia preprandial e hiperglicemia postprandial.
- Deficiencia de glucosa-6-fosfatasa (Glucogenosis tipo I), enzima que cataliza la conversión de G-6-P a glucosa, siendo la vía final común de la glucogenólisis y gluconeogénesis. Su carencia lleva a hipoglicemia severa, cetosis, acidosis láctica, hiperuricemia, hipertrigliceridemia, elevación de transaminasas y disfunción plaquetaria. El tipo Ia o enfermedad de Von Gierke da cuenta del 80-90% de los casos. El tipo Ib (defecto del transporte microsomal de la G-6-P), se acompaña de neutropenia real o funcional, haciendo a los individuos susceptibles a infecciones recurrentes; también se puede acompañar de enfermedad inflamatoria intestinal. Los niños que poseen este déficit enzimático presentan una facies descrita como de “muñeca”.
- Deficiencia de enzima desramificante (Glucogenosis tipo III o enfermedad de Cori); esta enzima rompe los enlaces 1-6 del glicógeno (actividad 1-6 glicosidasa) y transfiere los 3 últimos residuos de glucosa de una rama a la posición 1-4 del polímero lineal central de glucógeno (actividad 4-a-translocasa). Los niños con el déficit presentan hipoglicemia leve, hepatomegalia, retraso de crecimiento, cetosis, acidosis, hiperlipidemia, elevación de transaminasas y puede haber también compromiso cardíaco y muscular con elevación de CK.
- Deficiencia de fosforilasa hepática (glucogenosis tipo VI
o enfermedad de Hers), enzima que rompe los enlaces
1-4 del glicógeno, liberando glucosa-6-fosfato. Produce
hipoglicemia leve, hepatomegalia, retraso de crecimiento,
hiperlipidemia, cetosis, leve elevación de transaminasas.
En resumen, las glucogenosis se deben sospechar en pacientes que presenten hipoglicemia de ayuno, cetosis leve a moderada, retraso del crecimiento, con o sin hepatomegalia. El diagnóstico requiere biopsia hepática y se confirma con estudio molecular del defecto. El tratamiento se basa en alimentación frecuente y polímeros de glucosa de lenta liberación como es la maicena cruda5.
- Alteración de la gluconeogénesis: Se presentan con hipoglicemia cetótica y acidosis láctica ante el ayuno prolongado, luego de que se han depletado los depósitos de glicógeno. Las enzimas involucradas son la glucosa-6-fosfatasa, fructosa-1,6-difosfatasa, piruvato carboxilasa y fosfoenolpiruvatocarboxiquinasa (PEPCK).
Alteración del metabolismo de monosacáridos: Las formas más comunes son:
- Glactosemia: Deficiencia de galactosa-1-fosfato uridil
tranferasa. El cuadro clínico típico es hipoglicemia, diarrea
y vómitos luego de ingerir lactosa (glucosa + galactosa). La hipoglicemia se produce por acumulación de galactosa-1-fosfato que inhibe enzimas de la glucogenólisis y gluconeogénesis; además causa disfunción hepática y hepatomegalia, cataratas y retardo mental6. - Intolerancia hereditaria a la fructosa: Corresponde a deficiencia de fructosa-1-aldolasa que convierte fructosa- 1-fosfato en dihidroacetona fosfato y gliceraldehido. Se presenta cuando la fructosa o sucrosa (glucosa + fructosa) es incorporado a la dieta, causando vómitos e hipoglicemia postprandiales, acompañado de acidosis, hepatomegalia, ictericia e aminoaciduria.
Alteración del metabolismo de los aminoácidos
Este grupo comprende a las acidurias orgánicas, como enfermedad de orina con olor a jarabe de arce, acidemia propiónica, metilmalónica, isovalérica, glutárica y tirosinemia. Se presentan durante el período de recién nacido o en la infancia temprana, habitualmente con episodios de acidosis metabólica grave, con aumento del “anion gap”, lo que suele confundirse con sepsis. La hipoglicemia se relaciona con enfermedad hepática, malnutrición proteica y/o deficiencia de carnitina por pérdida renal.
Alteraciones del metabolismo de los ácidos grasos
Incluyen alteración en la b-oxidación, como deficiencia de SCHAD, MCAD LCHAD (acyl-CoA-deshidrogenasa de cadena corta, mediana y larga respectivamente), metabolismo y transporte de carnitina y formación de cuerpos cetónicos. La hipoglicemia ocurre luego de períodos prolongados de ayuno habitualmente relacionado a una enfermedad intercurrente. La hipoglicemia es grave, hipocetótica, con carnitina baja y ácidos grasos libres elevados. En general existe un importante compromiso de conciencia, hipotonía, hepatomegalia, elevación de enzimas hepáticas y musculares y puede agregarse falla cardíaca, rabdomiolisis y edema cerebral. El tratamiento consiste en administración de glucosa suficiente para aumentar la insulina y así suprimir la lipólisis y proteólisis; en estados catabólicos como trauma, quemaduras o infecciones deben recibir alimentación frecuente rica en carbohidratos para evitar la movilización endógena de sustratos7 y administración de carnitina cuando exista déficit.
La hipoglicemia cetótica
Se presenta entre 18 meses y 5 años de vida, siendo la causa más frecuente de hipoglicemia a esta edad; remite espontáneamente entre los 8 y 9 años. Se produce en niños delgados con poca masa muscular, que presentan hipoglicemia ante un ayuno más prolongado de lo normal, habitualmente cursando alguna enfermedad intercurrente. La hipoglicemia ocurre en el momento de transición entre glicogenólisis y neoglucogénesis, y su patogenia no está del todo clara, pero se piensa que tiene que ver con la disponibilidad y movilización de sustratos para la neoglucogénesis, especialmente alanina, proveniente del tejido muscular y con la disminución en la actividad de las enzimas involucradas en este proceso. También se ha postulado una utilización insuficiente de los cuerpos cetónicos o simplemente que sería el extremo de una curva de tolerancia normal al ayuno8.
El diagnóstico requiere descartar déficit hormonales y enfermedades hepáticas que también puedan causar hipoglicemia cetótica. El manejo consiste en evitar ayunos prolongados y en caso de enfermedades intercurrentes aportar soluciones glucosadas. Una forma de predecir la aparición de la hipoglicemia es medir cuerpos cetónicos en orina.
II. Deficiencias hormonales
Los estados deficitarios de hormona de crecimiento (GH) y cortisol pueden ser causa de hipoglicemia en todas las edades. Producen hipoglicemia por disminución de la proteinolisis (cortisol), disminución de la neoglucogénesis (GH y cortisol), y aumento de la sensibilidad periférica a la acción de la insulina ya que ambas hormonas antagonizan su acción. El déficit de hormonas tiroideas también podría producir hipoglicemia aunque el mecanismo no es claro. Se debe sospechar déficit de GH en el recién nacido que tiene asociado un micropene, microftalmia o un defecto de la línea media. El deterioro en la curva de crecimiento ocurre después de los 6 a 12 meses de edad. El déficit de cortisol puede ser producido por hiperplasia o hipoplasia suprarrenal congénita, enfermedad de Addison, deficiencia o resistencia a ACTH y se presenta con hipoglicemia, hipotensión postural, fatiga, anorexia, baja de peso, hiperpigmentación o alteraciones hidroelectrolíticas, crisis perdedora de sal y shock.
En el período neonatal, por la inmadurez enzimática, puede remedar al hiperinsulinismo con ácidos grasos libres y cuerpos cetónicos disminuidos, aunque en niños mayores los cuerpos cetónicos y los ácidos grasos libres están elevados. El diagnóstico se confirma con bajos niveles hormonales durante la hipoglicemia y el tratamiento es la suplementación hormonal.
III. Drogas
- Alcohol: Por disminución de la ingesta alimenticia, que lleva a depósitos bajos de glicógeno y disminución de la neoglucogénesis por aumento de la relación NADH/NAD, disminución de la liberación de alanina del músculo y de la captación hepática de sustratos neoglucogénicos. Se acompaña de acidosis metabólica con ácido láctico aumentado.
- Insulina: Por sobredosis en un paciente diabético o de forma intencional (Munchausen). En la muestra crítica la insulina alta y el péptido C bajo ayudan a establecer el diagnóstico.
- Sulfonilureas: Estimulan la liberación de insulina de la célula b; se debe investigar sobre posible acceso a esta droga en familiares.
- b-bloqueadores: Facilitan la hipoglicemia al bloquear la respuesta autonómica típica ante ella (estimulación de glicogenólisis y gluconeogénesis, inhibición de la utilización periférica y de la secreción insulínica); además inhiben los síntomas de la hipoglicemia llegando ésta a ser más profunda. Los ácidos grasos y cuerpos cetónicos están suprimidos.
- Salicilatos: Aumentan la utilización periférica de glucosa sumado al mecanismo utilizado por los b-bloqueadores. Clínicamente hay hiperventilación y depresión del sensorio y en el laboratorio se evidencia alcalosis respiratoria y acidosis metabólica.
IV. Hiperinsulinismo persistente
El hiperinsulinismo persistente o hiperinsulinismo congénito (HIC) es la principal causa de hipoglicemia persistente en el menor de un año9. Se caracteriza por una disregulación de la secreción insulínica que genera hipoglicemia severa. Por un lado bloquea la glicogenólisis, y por otro inhibe la neoglucogénesis, cetogénesis y lipólisis impidiendo que la glucosa sea sintetizada a partir de sustratos periféricos. Además, deja al SNC sin fuente alternativa de energía, produciendo frecuentemente convulsiones y secuelas neurológicas.
Los criterios diagnósticos incluyen hipoglicemia recurrente, tanto de ayuno como postprandial, necesidad de elevadas cargas de glucosa (> 10 mg/kg/min) para mantener glicemia normal, insulina detectable y ausencia de cuerpos cetónicos al momento de la hipoglicemia, con test de glucagón positivo.
En la regulación de la secreción insulínica participa el
canal de K+, ATP dependiente, en la membrana de la célula
b pancreática, el cual es un octámero compuesto por 4 subunidades
regulatorias SUR1, codificadas por el gen ABCC8 y
4 subunidades Kir 6.2, codificadas por el gen KCNJ11, que
conforman el poro central del canal. Ambos genes localizados
en el cromosoma 11p15.110. Este canal sensa la relación
ATP/ADP intracelular; al entrar glucosa a la célula y ser metabolizada,
aumenta esta relación, produciéndose el cierre
del canal, con aumento del K intracelular, depolarizándose la
membrana plasmática con entrada de calcio a la célula y con
la consiguiente liberación de insulina desde los gránulos de
depósito (Figura 2).
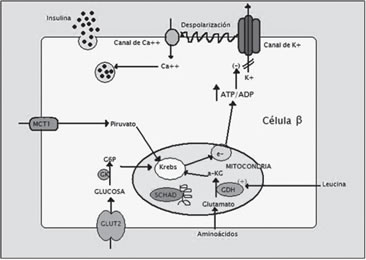 Figura 2. Esquema de regulación de la secreción insulínica
y mecanismos moleculares.
Glut2: transportador de glucosa 2; GK: glucokinasa;
SCHAD: acyl-CoA dehidrogenasa de cadena corta;
GDH: glutamato dehidrogenasa; e-: cadena respiratoria;
MCT1; trasportador de monocarboxilatos 1.
Figura 2. Esquema de regulación de la secreción insulínica
y mecanismos moleculares.
Glut2: transportador de glucosa 2; GK: glucokinasa;
SCHAD: acyl-CoA dehidrogenasa de cadena corta;
GDH: glutamato dehidrogenasa; e-: cadena respiratoria;
MCT1; trasportador de monocarboxilatos 1.
El hiperinsulinismo congénito se puede dividir en 2 grupos; las canalopatías en las que se compromete el canal de K+, ya sea estructural, funcional o regulatoriamente; ésta es la forma más frecuente de HIC (40-50%), (a la fecha se conocen más de 150 mutaciones inactivantes en ABCC8 y 10 en KCNJ1111). El segundo grupo lo constituyen las metabolopatías en que se altera la relación ATP/ADP intracelular y que incluyen mutaciones en las siguientes enzimas:
- Glucokinasa (gen GCK): Fosforila glucosa a glucosa-6- fosfato, actúa como sensor de glucosa, ya que la tasa de fosforilación es directamente proporcional a la glucosa plasmática. Mutaciones activantes aumentan su afinidad por glucosa alterando el umbral de liberación de insulina mediado por glucosa.
- Glutamato dehidrogenasa (gen GLUD1): Cataliza la deaminación oxidativa de glutamato a amonio más acetoglutarato el que entra al ciclo de Krebs para producir ATP. Mutaciones activantes producen hiperinsulinismo e hiperamonemia. Es la segunda causa más frecuente de HIC. Se caracteriza por hipoglicemia desencadenada por la ingesta proteica, hiperamonemia y convulsiones independientes de la hipoglicemia12. El tratamiento consiste en restricción proteica, diazoxide y en ocasiones benzoato de sodio o N-carbamilglutamato.
- Hidroxiacyl CoA dehidrogenasa de cadena corta (SCHAD): Cataliza el penúltimo paso de b-oxidación de ácidos grasos: de 3-hidroxiacylCoA a 3-cetoacylCoA. El mecanismo por el cual produce hiperinsulinismo no está del todo claro, pero parece ser que esta enzima es un regulador negativo de la secreción de insulina, de tal manera que una pérdida de función genera hiperinsulinismo13. La forma de presentación va desde hipoglicemia neonatal hasta formas leves tardías; la muestra crítica revela aumento del 3 hidroxiglutarato en orina e hidroxibutyrylcarnitina en sangre.
- Factor hepatocítico nuclear 4a (HNF4a): Las mutaciones heterocigotas dominantes producen hipoglicemia, hiperinsulinemica transitoria y se caracterizan por un alto peso de nacimiento con desarrollo posterior de diabetes MODY-114. El mecanismo del hiperinsulinismo es poco claro.
- SCL16A1: gen que codifica para MCT1, transportador de
monocarboxilatos como lactato y piruvato. Un aumento
en la expresión de MCT1 produce típicamente hipoglicemia
dentro de los 30 minutos siguientes a un corto período
de ejercicio anaeróbico, debido a aumento del lactato
y piruvato que entra al ciclo de Krebs, aumentando la
relación ATP/ADP y secretando insulina15.
Existen además hiperinsulinismos asociados a síndromes genéticos como Beckwith Wiedemann, Sotos, Perlman, Simpson-Golabi-Behmel, Kabuki, Fanconi-Bickel, Usher, Timothy, Costello, Trisomia 13, Turner y otros asociados a condiciones metabólicas congénitas como desordenes de glicosilación 1ª/b/d , citrulinemia, deficiencia de complejo III y IV de cadenas respiratorias y la Tirosinemia tipo 1. También hay que mencionar el hiperinsulinismo persistente del PEG, el cual no es infrecuente; aunque su mecanismo se desconoce, responden bien al diazoxide y su evolución es hacia la remisión espontánea (entre < 1 y 10 meses de edad)16.
Histología del Hiperinsulinismo Congénito
El HIC se divide histológicamente en 2 formas, focal y difusa. La hiperplasia adenomatosa focal se compone de pequeñas agrupaciones de islotes pancreáticos hiperplásicos separados por unos pocos acinos exocrinos, manteniendo la normal arquitectura lobular pancreática. Estas lesiones focales son distintas a los insulinomas, tanto en su histología como en los mecanismos moleculares de la secreción insulínica. La forma difusa se caracteriza por células con abundante citoplasma y gran núcleo en los islotes a través de todo el páncreas17. La diferenciación entre focal y difuso es crucial para decidir el tratamiento, ya que en el caso de ser focal, la resección quirúrgica es curativa, mientras que si es difuso requiere pancreatectomía subtotal con tasas de curación que son variables y con riesgo de disfunción pancreática exocrina y desarrollo de diabetes en forma tardía18.
Ambas formas pueden ser indistinguibles en cuanto a su presentación clínica. La localización es importante cuando el paciente no responde a terapia oral con diaxozide y se requiere intervención quirúrgica. Desafortunadamente los exámenes imagenológicos disponibles en Chile (ecografía, TAC, RN) no nos permiten diferenciarlas. El cateterismo transhepático pancreático con muestreo venoso de las diferentes afluentes pancreáticas o la estimulación intrarterial con calcio para ver la respuesta insulínica aguda son procedimientos que se utilizaron para diferenciar la forma focal de la difusa, pero son invasivos y de alta complejidad. Recientemente, el examen de elección es la tomografía de emisión de positrones con 18-fluoro-dihidroxifenilalanina (18 F-DOPA-PET), que ha permitido una mejor localización preoperatoria, describiéndose una precisión de 88%, valor VPP de 100% y VPN de 81% en 50 niños con HIC19. Este examen no esta, a la fecha, disponible en Chile.
Afortunadamente, existe una buena correlación histopatológica con el genotipo20 y la respuesta a tratamiento. La forma focal se debe a mutación germinal del alelo paterno ABCC8/KCNJ11 más deleción somática del alelo materno21 produciéndose un desbalance entre el locus H19 (supresor de tumor) e IGF-II (promotor de crecimiento). La forma difusa se hereda en forma autosómica recesiva con mutaciones en ABCC8 o KCNJ11, o menos frecuentemente en forma autosómica dominante de GCK o GLUD122.
Tratamiento del Hiperinsulinismo Congénito
El diazoxide (5-20 mg/kg/d dividido en 2 dosis) es el tratamiento de primera línea una vez establecido el diagnóstico de hiperinsulinismo. Es de uso oral y de bajo costo y actúa abriendo el canal de K+, inhibiendo la liberación de insulina23, y se recomienda administrarlo junto con hidroclotiazida (7-10 mg/kg/d en 2 dosis) que potencia su acción y evita retención hídrica. Otros efectos adversos del diazoxide son hipertricosis, discrasias sanguíneas y disminución de IgA. La respuesta al tratamiento es variable siendo mayor en la formas autosómicas dominantes como mutaciones de GCK y GLUD1 y menor en las recesivas como mutaciones de KCNJ11 o ABCC820.
El Nifedipino (0,25-2,5 mg/kg/d en 3 dosis) bloquea los canales de calcio dependientes de voltaje, disminuyendo la secreción insulínica, pero, en general, la respuesta es pobre y su dosificación difícil en los niños pequeños.
De segunda línea son medicamentos de uso parenteral como octreotide y glucagón, generalmente administrados en forma transitoria para la estabilización previa a la pancreatectomía, existiendo pocas comunicaciones de su uso en forma prolongada24,25. El glucagón tiene efecto al movilizar los depósitos de glicógeno hepático. Se puede administrar vía infusión ev continua (1-10 mg/kg/h). Por ser un potente secretagogo de insulina, se propone usarlo en forma conjunta al octeotride (5-20 mg/kg/d ev o sc) para evitar hipoglicemia de rebote.
El octreotide induce hiperpolarización de la célula impidiendo la liberación de insulina. Es necesario controlar eventuales efectos adversos como intolerancia gastrointestinal, vómitos, diarrea, distensión abdominal, malabsorción, colelitiasis, así como inhibición de la secreción de otras hormonas como GH26, TSH, glucagón, gastrina, VIP y otras27.
La buena respuesta al tratamiento se define como un régimen aceptable para la familia, y con glicemias normales durante un período de ayuno razonable.
El consenso actual es que aquellos niños con hiperinsulinismo de inicio precoz, en que se ha intentado manejo médico agresivo por 4-6 semanas sin buena respuesta, debieran ser evaluados con PET scan para decidir luego una cirugía focal o total de acuerdo a la lesión encontrada.
Existen pocas series publicadas con respecto al tratamiento quirúrgico del HIC. En un grupo de 114 HIC en que se realizó pancreatectomía a 60, se logró euglicemia en sólo 27% de los casos, requiriendo reintervención por recurrencia de hipoglicemia el 28,3%, de los cuales el 71% desarrollaron DM en un seguimiento promedio de 6,6 años, presentándose ésta mayoritariamente dentro del primer año después de la cirugía o durante la pubertad18 La función exocrina del páncreas también suele verse afectada después de la pancreatectomía subtotal, requiriendo ser monitorizada con elastasa fecal.
La historia natural del HIC ha sido descrita en algunos pacientes que se han controlado médicamente, demostrando que desarrollan a largo plazo intolerancia a la glucosa o diabetes mellitus28,29. El mecanismo no está del todo aclarado pero se postula que existiría una progresiva apoptosis de la célula b dada por el stress metabólico causado por la continua secreción de insulina30.
Conclusiones
Es importante considerar que la hipoglicemia es un signo y no un diagnóstico, por lo tanto, es fundamentan encontrar la causa de ella, para poder disponer las medidas adecuadas de tratamiento y evitar su recurrencia, de modo de prevenir un daño neurológico futuro. Los avances en el estudio molecular nos permiten en muchos casos diagnosticar con certeza la causa de la hipoglicemia y tomar conductas terapéuticas específicas, así como también proporcionar un adecuado consejo genético.
Referencias
- Burns CM, Rutherford MA, Boardman JP, Cowan FM. 2008. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics 122 (1): 65-74.
- Cornblath M, Hawdon JM, Williams AF, Aynsley-Green A, Ward- Platt MP, Schwartz R, Kalhan SC. 2000. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics 105 (5): 1141-1145.
- Koh TH, Aynsley-Green A, Tarbit M, Eyre JA. 1988. Neural
dysfunction during hypoglycaemia. Arch Dis Child 63 (11):
1353-1358. - Basu P, Som S, Choudhuri N, Das H. 2009. Contribution of the blood glucose level in perinatal asphyxia. Eur J Pediatr 168: 833-838.
- Heller S, Worona L, Consuelo A. 2008. Nutritional therapy for glycogen storage diseases. J Pediatr Gastroenterol Nutr 47 Suppl 1: S15-21.
- Bosch AM. 2006. Classical galactosaemia revisited. J Inherit Metab Dis 29 (4): 516-525.
- Morris AA, Leonard JV. 1997. Early recognition of metabolic decompensation. Arch Dis Child 76 (6): 555-556.
- Huidekoper HH, Duran M, Turkenburg M, Ackermans MT,
Sauerwein HP, Wijburg FA. 2008. Fasting adaptation in idiopathic
ketotic hypoglycemia: a mismatch between glucose production and
demand. Eur J Pediatr 167 (8): 859-865.
- Stanley CA. 1997 Hyperinsulinism in infants and children. Pediatr Clin North A 44: 363-374.
- Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J,
González G, et al. 1995. Reconstitution of KATP: An inward
rectifier subunit plus the sulphonylurea receptor. Science 270: 1166-1170. - Flanagan SE, Clauin S, Bellanne-Chantelot C, et al. 2009 Update of mutations in the genes encoding the pancreatic beta-cell K (ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat 30: 170-180.
- Kapoor R, Flanagan S, Fulton P, et al. 2009. Hyperinsulinismhyperammonaemia
(HI/HA) syndrome: novel mutations in
the GLUD1 gene and genotypephenotype correlations. Eur J Endocrinol 161: 731-735. - Hardy OT, Hohmeier HE, Becker TC, et al. 2007. Functional genomics of the betacell:short-chain 3-hydroxyacyl-coenzyme A dehydrogenase regulates insulin secretion independent of Kþ currents. Mol Endocrinol 21: 765-773.
- Conn JJ, Simm PJ, Oats JJ, et al. 2009. Neonatal hyperinsulinaemic hypoglycaemia and monogenic diabetes due to a heterozygous mutation of the HNF4A gene. J Obstet Gynaecol 49: 328-330.
- Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, et al. 2007. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet 81 (3): 467-474.
- Hoe FM, Thornton PS, Wanner LA, Steinkrauss L, Simmons RA, Stanley CA. 2006. Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J Pediatr 148 (2): 207-212.
- Kloppel G, Reinecke-Luthge A, Koschoreck F. 1999. Focal and diffuse b-cell changes in persistent hyperinsulinemic hypoglycemia of infancy. Endocr Pathol 10: 299-304.
- Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. 2003. Long-term follow-up of 114 patiens with congenital hyperinsulinism. Eur J Endocr 149: 43-51.
- Hardy OT, Hernández-Pampaloni M, Saffer JR, Scheuermann JS, Ernst LM, Freifelder R, et al. 2007. Accuracy of [18 F] fluorodopa positron emission tomography for diagnosing and localizing focal congenital hyperinsulinism. J Clin Endocrinol Metab 92: 4706-4711.
- Kapoor R, James C, Hussain K. 2009 Advances in the diagnosis and management of hyperinsulinaemic hypoglycaemia. Nat Clin Pract Endocrinol Metab 5: 101-112.
- Verkarre V, Fournet JC, De Lonlay P, Gross-Morand MS, Devillers M, Rahier J, Brunelle F, Robert JJ, Nihoul-Fékété C, Saudubray JM, Junien C. 1998. Paternal Mutation of the Sulfonylurea Receptor (SUR1) Gene and Maternal Loss of 11p15 Imprinted Genes Lead to Persistent Hyperinsulinism in Focal Adenomatous Hyperplasia. J Clin Invest; 102: 1286-1291.
- Giurgea I, Bellanné-Chantelot C, Ribeiro M, Hubert L, Sempoux C, Robert JJ, et al. 2006. Molecular mechanisms of neonatal hyperinsulinism. Horm Res 66: 289-296.
- Hussain K. 2008. Diagnosis and management of hyperinsulinaemic hypoglycaemia of infancy. Horm Res 69: 2-13.
- Glaser B, Hirsch HJ, Landau H. 1993. Persistent hyperinsulinemic hypoglycemia of infancy: long-term octreotide treatment without pancreatectomy. J Pediatr 123 (4): 644-650.
- Mohnike K, Blankenstein O, Pfuetzner A, Pötzsch S, Schober E, Steiner S, et al. 2008. Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Res Horm 70: 59-64.
- Thornton PS, Alter CA, Levitt KLE, Baker L, Stanley CA. 1993. Short and long-term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr 123: 637-643.
- Tauber MT, Harris AG, Rochiccioli P. 1994. Clinical use of the long acting somatostatin analogue octreotide in pediatrics. Eur J Pediatr 153: 304-310.
- Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, Perry A, Abu-Libdeh A, Glaser B, et al. 2008. Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes 57 (7): 1935-1940.
- Gussinyer M, Clemente M, Cebrián R, Yeste D, Albisu M, Carrascosa A. 2008. Glucose intolerance and diabetes are observed in the long-term follow-up of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care 31: 1257-1259.
- Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. 2000. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49: 1325-1333.